Tibor I. Krisko, Ehrin J. Armstrong, and David E. Cohen
BIOCHEMISTRY AND PHYSIOLOGY OF CHOLESTEROL AND LIPOPROTEIN METABOLISM |

Lipids are insoluble or sparingly soluble molecules that are essential for membrane biogenesis and maintenance of membrane integrity. They also serve as energy sources, hormone precursors, and signaling molecules. In order to facilitate transport through the relatively aqueous environment of the blood, nonpolar lipids, such as cholesteryl esters and triglycerides, are packaged within lipoproteins.
Increased concentrations of certain lipoproteins in the circulation are associated strongly with atherosclerosis. Much of the prevalence of cardiovascular disease (CVD), the leading cause of death in the United States and most Western countries, can be attributed to elevated blood concentrations of cholesterol-rich low-density lipoprotein (LDL) particles as well as lipoproteins that are rich in triglycerides. Epidemiologically, decreased concentrations of high-density lipoproteins (HDL) also predispose to atherosclerotic disease. The major contributors to lipoprotein abnormalities appear to be Western diets combined with sedentary lifestyles, but a limited number of genetic causes of hyperlipidemia have also been identified. The role of genetics in common forms of hyperlipidemia is the subject of intense study utilizing cutting-edge genomic approaches. It is apparent that genes modify both the sensitivity of individuals to adverse dietary habits and lifestyles and the response of individuals to lipid-lowering therapies.
This chapter highlights the biochemistry and physiology of cholesterol and lipoproteins, with an emphasis on the role of lipoproteins in atherogenesis, and the pharmacologic interventions that can ameliorate hyperlipidemia. Abundant clinical outcomes data have proven that morbidity and mortality from cardiovascular disease can be reduced by the use of lipid-lowering drugs.
 Jake P, a 29-year-old construction worker, makes an appointment to see Dr. Cush. Jake complains of hard, elevated swellings around his Achilles tendon that seem to rub constantly against his construction boots. Jake had been hesitant to see the doctor (his last appointment was 10 years ago), but he remembers that his dad, who died at age 42 of a heart attack, had similar swellings. On examination, Dr. Cush recognizes the Achilles swellings as xanthomas (lipid deposits); the physical exam is otherwise within normal limits. Jake comments that his diet is quite “fatty,” including three to four donuts each day and frequent hamburgers. Dr. Cush explains that the xanthomas on Jake’s feet are the result of cholesteryl ester deposition, probably from high cholesterol levels in his blood. Dr. Cush orders a fasting plasma cholesterol level and recommends that Jake reduce his intake of foods high in saturated fat and cholesterol and increase his intake of poultry, fish, whole cereal grains, fruits, and vegetables. Jake has gained about 15 pounds since he was 19 and has a small paunch. Dr. Cush recommends regular exercise and weight loss.
Jake P, a 29-year-old construction worker, makes an appointment to see Dr. Cush. Jake complains of hard, elevated swellings around his Achilles tendon that seem to rub constantly against his construction boots. Jake had been hesitant to see the doctor (his last appointment was 10 years ago), but he remembers that his dad, who died at age 42 of a heart attack, had similar swellings. On examination, Dr. Cush recognizes the Achilles swellings as xanthomas (lipid deposits); the physical exam is otherwise within normal limits. Jake comments that his diet is quite “fatty,” including three to four donuts each day and frequent hamburgers. Dr. Cush explains that the xanthomas on Jake’s feet are the result of cholesteryl ester deposition, probably from high cholesterol levels in his blood. Dr. Cush orders a fasting plasma cholesterol level and recommends that Jake reduce his intake of foods high in saturated fat and cholesterol and increase his intake of poultry, fish, whole cereal grains, fruits, and vegetables. Jake has gained about 15 pounds since he was 19 and has a small paunch. Dr. Cush recommends regular exercise and weight loss.
Results of the blood test reveal a total plasma cholesterol concentration of 315 mg/dL (normal, <200), with elevated LDL cholesterol of 250 mg/dL (desirable, <100), low HDL of 35 mg/dL (normal, 35 to 100), and normal concentrations of triglycerides and very-low-density lipoprotein (VLDL). Based on these test results, his age, the Achilles heel xanthomas, and a positive family history for an early myocardial infarction, Dr. Cush tells Jake that he likely has an inherited disorder of cholesterol metabolism known as heterozygous familial hypercholesterolemia. This disease puts Jake at very high risk for early atherosclerosis and myocardial infarction. The low HDL cholesterol level also contributes to his increased risk of cardiovascular disease.
Dr. Cush tells Jake that aggressive lowering of cholesterol levels can ameliorate many of the disease sequelae. In addition to the dietary changes, Dr. Cush prescribes a statin to help reduce Jake’s cholesterol. A starting dose of a statin reduces his LDL by 45% to 138 mg/dL, while his HDL increases slightly. Dr. Cush then increases the statin dose, and this produces an additional 12% reduction in LDL. Because LDL has still not reached <100 mg/dL, and HDL remains low, Dr. Cush adds the cholesterol absorption inhibitor ezetimibe as well as extended-release niacin. After these modifications, Jake’s LDL drops below 100, and his HDL increases to 45 mg/dL. Jake experiences cutaneous flushing during the first few months of niacin treatment, but after that period, he has only occasional flushing episodes.
Questions
1. How do high cholesterol levels predispose to cardiovascular disease?
2. What is the etiology of familial hypercholesterolemia?
3. How do statins, ezetimibe, niacin, lomitapide, mipomersen, and PCSK9 inhibitors act pharmacologically?
4. What are the major adverse effects of concomitant statin and niacin therapy about which Jake should be aware?
 BIOCHEMISTRY AND PHYSIOLOGY OF CHOLESTEROL AND LIPOPROTEIN METABOLISM
BIOCHEMISTRY AND PHYSIOLOGY OF CHOLESTEROL AND LIPOPROTEIN METABOLISM
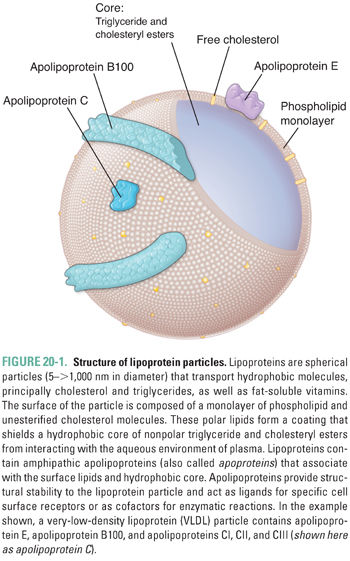
Lipoproteins are macromolecular aggregates that transport triglycerides and cholesterol in the blood. Circulating lipoproteins can be differentiated on the basis of density, size, and protein content (Table 20-1). As a general rule, larger, less dense lipoproteins have a higher percentage composition of lipids; chylomicrons are the largest and least dense lipoprotein subclass, whereas HDLs are the smallest lipoproteins, containing the lowest lipid content and the highest proportion of protein.
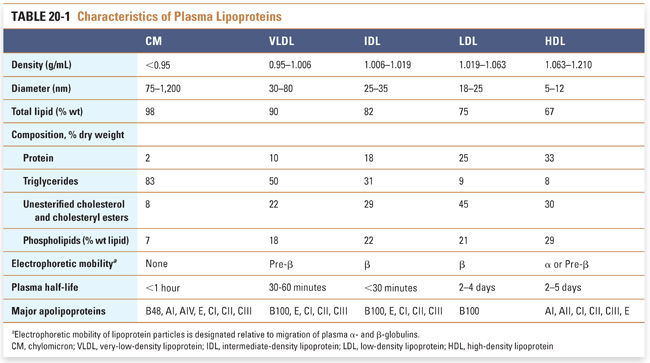
Structurally, lipoproteins are microscopic spherical particles ranging from 5 to >1,000 nm in diameter. Each lipoprotein particle consists of a monolayer of polar, amphipathic lipids that surrounds a hydrophobic core. Each lipoprotein particle also contains one or more types of apolipoprotein (Fig. 20-1). The polar lipids that comprise the surface coat are unesterified cholesterol and phospholipid molecules arranged in a monolayer. The hydrophobic core of a lipoprotein contains cholesteryl esters (cholesterol molecules linked by an ester bond to a fatty acid) and triglycerides (three fatty acids esterified to a glycerol molecule). Apolipoproteins (also referred to as apoproteins) are amphipathic proteins that intercalate into the surface coat of lipoproteins. In addition to stabilizing the structure of lipoproteins, apolipoproteins engage in biological functions. They may act as ligands for lipoprotein receptors or may activate enzymatic activities in the plasma. The apolipoprotein composition determines the metabolic fate of the lipoprotein. For example, each LDL particle contains one apolipoprotein B (apoB) 100 molecule, which is a ligand for the low-density lipoprotein receptor (discussed below); in turn, binding of LDL to the LDL receptor promotes cholesterol uptake into cells.
From a metabolic perspective, lipoprotein particles can be divided into lipoproteins that participate in the delivery of triglyceride molecules to muscle and fat tissue (the apoB-containing lipoproteins, chylomicrons, and VLDL) and lipoproteins that are involved primarily in cholesterol transport (HDL and the remnants of apoB-containing lipoproteins). HDL also serves as a reservoir for exchangeable apolipoproteins in the plasma, including apoAI, apoCII, and apoE. The following discussion presents each lipoprotein class in the context of its function.
Metabolism of ApoB-Containing Lipoproteins
The primary function of apoB-containing lipoproteins is to deliver fatty acids in the form of triglycerides to muscle tissue for use in ATP biogenesis and to adipose tissue for storage. Chylomicrons are formed in the intestine and transport dietary triglycerides, whereas VLDL particles are formed in the liver and transport triglycerides that are synthesized endogenously. The metabolic lifespan of apoB-containing lipoproteins can be divided into three phases: assembly, intravascular metabolism, and receptor-mediated clearance. This is a convenient categorization because pharmacologic agents are available that influence each phase.
Assembly of ApoB-Containing Lipoproteins
The cellular mechanisms by which chylomicrons and VLDL are assembled are quite similar. Regulation of the assembly process depends on the availability of apoB and triglycerides, as well as the activity of microsomal triglyceride-transfer protein (MTP).
The gene that encodes apoB is transcribed principally in the intestine and the liver. Apart from this tissue-specific expression, there is little transcriptional regulation of the apoB gene. In contrast, a key regulatory event that differentiates chylomicron metabolism from VLDL metabolism is the editing of apoB mRNA (Fig. 20-2). Within enterocytes but not hepatocytes, a protein named apoB editing complex-1 (apobec-1) is expressed. This protein constitutes the catalytic subunit of the apoB editing complex, which deaminates a cytosine at position 6666 of the apoB mRNA molecule. Deamination converts the cytosine to uridine. As a result, the codon containing this nucleotide is converted from glutamine to a premature stop codon. When translated, the intestinal form apoB48 is 48% as long as the full-length protein that is expressed in the liver and referred to as apoB100. As a consequence, chylomicrons, the apoB-containing lipoprotein produced by the intestine, contain apoB48, whereas VLDL particles produced by the liver contain apoB100.
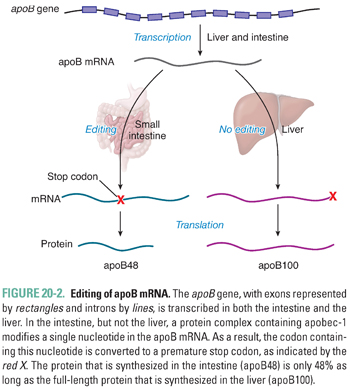
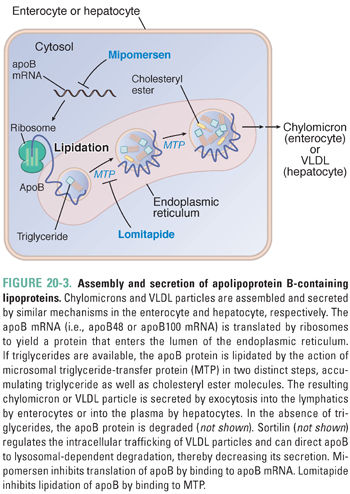
Figure 20-3 illustrates the cellular mechanisms by which apoB-containing lipoproteins are assembled and secreted. As the apoB protein is synthesized by ribosomes, it crosses into the endoplasmic reticulum. Within the endoplasmic reticulum, triglyceride molecules are added co-translationally to the elongating apoB protein (i.e., apoB is lipidated) by the action of a cofactor protein, MTP. Once apoB has been fully synthesized, the nascent lipoprotein is enlarged in the Golgi apparatus; during this process, MTP adds additional triglycerides to the core of the particle. By unclear mechanisms, cholesteryl esters are also added to the core. Each lipoprotein particle assembled by this process contains a single molecule of apoB.
Because the triglyceride component of chylomicrons originates primarily from the diet (Fig. 20-4), the assembly, secretion, and metabolism of chylomicrons are collectively referred to as the exogenous pathway of lipoprotein metabolism. During digestion, cholesteryl esters and triglycerides in food are hydrolyzed to form unesterified cholesterol, free fatty acids, and monoglycerides. Bile acids, phospholipids, and cholesterol are secreted by the liver into bile and stored in the gallbladder during fasting as micelles and vesicles, which are macromolecular lipid aggregates that form due to the detergent properties of bile acid molecules. The stimulus of eating a meal promotes emptying of gallbladder bile into the small intestine, where the micelles and vesicles solubilize the digested lipids.
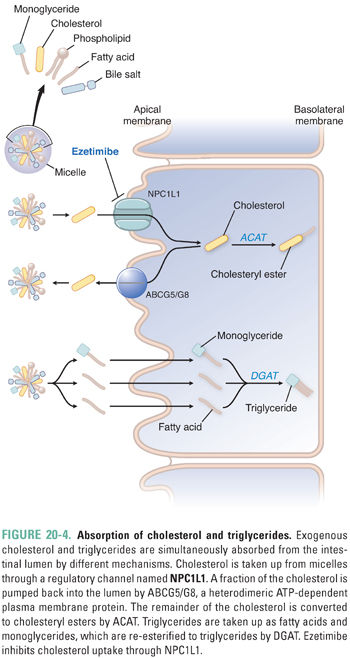
Lipid absorption into enterocytes of the duodenum and jejunum is facilitated mainly by micelles. Long-chain fatty acids and monoglycerides are taken up separately into the enterocyte by carrier-mediated transport and then re-esterified to form triglycerides by the enzyme diacylglycerol acyltransferase (DGAT). By contrast, medium-chain fatty acids are absorbed directly into the portal blood and metabolized by the liver. Dietary and biliary cholesterol from micelles enter the enterocyte via a protein channel named Niemann-Pick C1-like 1 protein (NPC1L1). Some of this cholesterol is immediately pumped back into the intestinal lumen by the ATP-dependent action of a heterodimeric protein, ABCG5/ABCG8 (ABCG5/G8). The fraction of cholesterol that remains is esterified to a long-chain fatty acid by acetyl-CoA:cholesterol acyltransferase (ACAT). Once triglycerides and cholesteryl esters are packaged together with apoB48, apoA1 is added as an additional structural apolipoprotein and the chylomicron particle is exocytosed into the lymphatics for transport to the circulation via the thoracic duct. The plasma concentration of triglyceride-rich chylomicrons varies in proportion to dietary fat intake.
Very-low-density lipoproteins (VLDL) contain triglycerides that are assembled by the liver using plasma fatty acids derived from adipose tissue or synthesized de novo. For this reason, the assembly, secretion, and metabolism of VLDL are often referred to as the endogenous pathway of lipoprotein metabolism. Hepatocytes synthesize triglycerides in response to increased free fatty acid flux to the liver. This typically occurs in response to fasting, thereby ensuring a continuous supply of fatty acids for delivery to muscle in the absence of triglycerides from the diet. Interestingly, dietary saturated fats as well as carbohydrates also stimulate the synthesis of triglycerides within the liver. By cellular mechanisms that are similar to those that produce chylomicrons (Fig. 20-3), MTP in hepatocytes lipidates apoB100 to form nascent VLDL particles. Under the continued influence of MTP, the nascent VLDL particles coalesce with larger triglyceride droplets and are secreted directly into the circulation. VLDL particles may also acquire apoE, apoCI, apoCII, and apoCIII within the hepatocyte prior to secretion. However, these apolipoproteins may also be transferred to VLDL from HDL in the circulation.
The synthesis of apoB48 in the intestine and apoB100 in the liver is constitutive. This permits the immediate production of chylomicrons and VLDL particles when triglyceride molecules are available. In the absence of triglycerides, such as in enterocytes during fasting, apoB is degraded by a variety of cellular mechanisms. Recent studies have revealed a role for sortilin in the cellular trafficking of VLDL particles. Sortilin is encoded by the Sort1 gene, and genome-wide association studies (GWAS) have shown that Sort1 is associated with reduced levels of LDL cholesterol (LDL-C). Sortilin facilitates the post-translational degradation of apoB by a lysosome-dependent mechanism.
Intravascular Metabolism of ApoB-Containing Lipoproteins
Within the circulation, chylomicrons and VLDL particles must be activated in order to target triglyceride delivery to muscle and adipose tissue (Fig. 20-5). Activation requires the addition of an optimal complement of apoCII molecules, which occurs by aqueous transfer of apoCII from HDL particles. Because there is an inherent delay in the transfer of apoCII to chylomicrons and VLDL particles, there is time for widespread circulation of triglyceride-rich particles throughout the body.
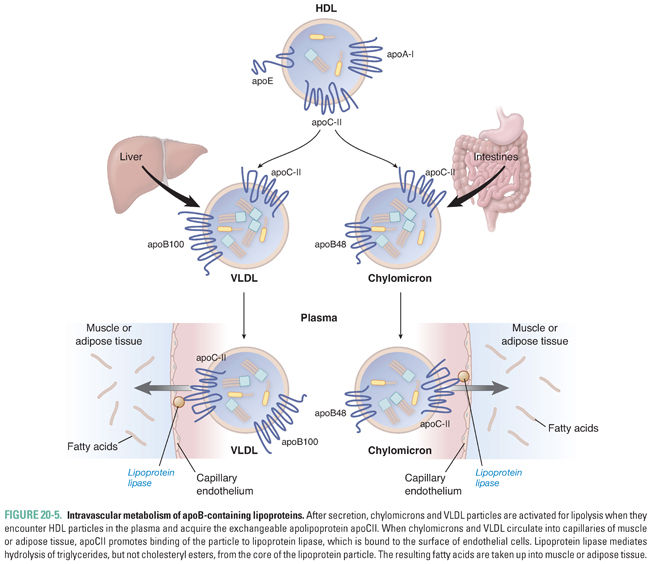
Lipoprotein lipase (LPL) is a lipolytic enzyme expressed on the endothelial surface of capillaries in muscle and fat tissue. LPL is a glycoprotein that is synthesized by myocytes and adipocytes and transported to the endothelial cell surface by a specific glycosylphosphatidylinositol (GPI)-linked protein, GPIHBP1. On the endothelial cell membrane, GPIHBP1 also serves to anchor LPL in place. Once chylomicrons and VLDL particles acquire apoCII, they can bind to LPL, which hydrolyzes triglycerides from the core of the lipoprotein (Fig. 20-5). LPL-mediated lipolysis liberates free fatty acids and glycerol. The free fatty acids are then taken up by the neighboring parenchymal cells. The expression level and intrinsic activity of LPL in muscle and adipose tissue are regulated according to the fed/fasting state, allowing the body to direct the delivery of fatty acids preferentially to muscle during fasting and to adipose after a meal. The rate of lipolysis of chylomicron and VLDL triglycerides is also controlled by apoCIII, which is an inhibitor of LPL activity. LPL inhibition by apoCIII may be an additional mechanism promoting widespread distribution of triglyceride-rich particles in the circulation.
Receptor-Mediated Clearance of ApoB-Containing Lipoproteins
As LPL continues to hydrolyze triglycerides from chylomicrons and VLDL, the particles become progressively depleted of triglycerides and relatively enriched in cholesterol. Once approximately 50% of the triglycerides have been removed, the particles lose their affinity for LPL and dissociate from the enzyme. The exchangeable apolipoproteins apoAI and apoCII (as well as apoCI and apoCIII) are then transferred to HDL in exchange for apoE (Fig. 20-6A), which serves as a high-affinity ligand for receptor-mediated clearance of the particles. Upon acquiring apoE, the particles are termed chylomicron or VLDL remnants.
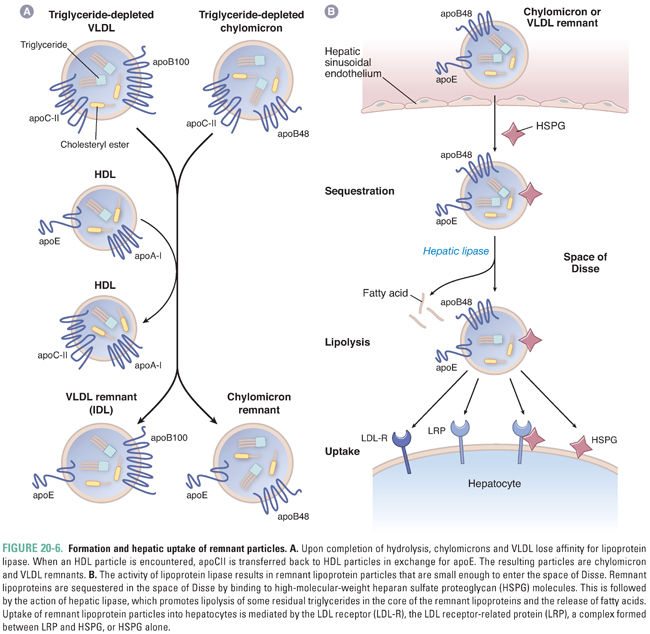
Remnants of chylomicrons and VLDL are taken up by the liver in a three-step process (Fig. 20-6B). The first step is sequestration of the particles within the space of Disse between the fenestrated endothelium of the liver sinusoids and the sinusoidal (basolateral) plasma membrane of the hepatocytes. Sequestration requires that the remnant particles become small enough during lipolysis to fit between the endothelial cells. Once in the space of Disse, remnants are bound and sequestered by large heparan sulfate proteoglycans. The next step is particle remodeling within the space of Disse by the action of hepatic lipase, a lipolytic enzyme that is similar to LPL but is expressed by hepatocytes. Hepatic lipase appears to optimize the triglyceride content of remnant particles so that they can be cleared efficiently by receptor-mediated mechanisms. The final phase of remnant clearance is receptor-mediated particle uptake. This is accomplished by one of four pathways. At the sinusoidal hepatocyte plasma membrane, remnant particles may be bound and taken up by the LDL receptor, the LDL receptor-related protein (LRP), or heparan sulfate proteoglycans. A fourth pathway is mediated by the combined activities of LRP and heparan sulfate proteoglycans. These redundant mechanisms allow for efficient particle clearance, so that the half-life of remnants in the plasma is approximately 30 minutes.
Formation and Clearance of LDL Particles
ApoB48-containing chylomicron remnants are completely cleared from the plasma. By contrast, the presence of apoB100 alters the metabolism of VLDL remnants so that only approximately 50% are cleared by the pathways for remnant particles. The difference is manifested during the metabolism of the remnant particles by LPL. VLDL remnants are avidly metabolized by LPL, becoming an increment smaller, relatively more deficient in triglycerides, and relatively enriched in cholesteryl esters. When converted to remnants following exchange of apolipoproteins with HDL, these more dense particles are called intermediate-density lipoproteins (IDL). Because IDL contain apoE, a fraction of these particles (approximately 50%) may be cleared by the liver via remnant receptor pathways (Fig. 20-6). However, the remainder are converted to LDL by hepatic lipase, which further hydrolyzes triglycerides in the core of IDL. The further reduction in size of the particle results in the transfer of apoE to HDL. As a consequence, LDL is a distinct, cholesteryl ester-enriched lipoprotein with apoB100 as its only apolipoprotein (Fig. 20-7A).
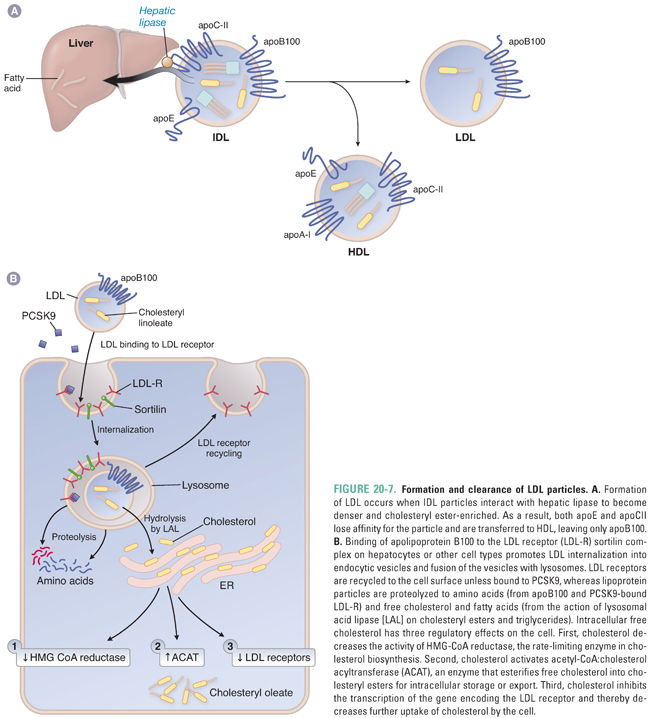
The LDL receptor is the only receptor capable of clearing significant amounts of LDL from the plasma. The LDL receptor is expressed on the surface of hepatocytes, macrophages, lymphocytes, adrenocortical cells, gonadal cells, and smooth muscle cells. Due to the lack of apoE, LDL particles are relatively weak ligands for the LDL receptor. As a result, the half-life of LDL in the circulation is markedly prolonged (2–4 days). This explains why LDL cholesterol accounts for approximately 65–75% of total plasma cholesterol. The uptake of LDL-C by the LDL receptor also appears to be promoted by sortilin (Fig. 20-7B).
Interaction of apoB100 with the LDL receptor facilitates receptor-mediated endocytosis of LDL particles and subsequent vesicle fusion with lysosomes (Fig. 20-7B). The LDL receptor is recycled to the cell surface, while the cholesteryl esters and triglycerides within the LDL particle are hydrolyzed by lysosomal acid lipase (LAL) to release unesterified cholesterol and fatty acids. These hydrolysis products affect three major homeostatic pathways. First, intracellular cholesterol inhibits HMG-CoA reductase, the enzyme that catalyzes the rate-limiting step in de novo cholesterol synthesis. Second, cholesterol activates ACAT to increase esterification and storage of cholesterol in the cell. Third, LDL receptor expression is down-regulated, reducing further uptake of cholesterol into the cells. The majority of LDL receptors (70%) are expressed on the surface of hepatocytes. As a result, the liver is primarily responsible for the removal of LDL particles from the circulation.
Proprotein convertase subtilisin-like kexin type 9 (PCSK9) is a plasma protein that regulates LDL receptor activity. PCSK9 is synthesized as a 72-kDa proPCSK9 proprotein that is autocatalytically cleaved in the endoplasmic reticulum to form the mature protein. It then enters the secretory pathway, and with the assistance of sortilin in the trans-Golgi network, it is secreted into the plasma. PCSK9 then binds to the epidermal growth factor-like repeat A (EGF-A) motif of the LDL receptor. This complex is targeted to lysosomes for degradation (Fig. 20-7B). Gain-of-function mutations in PCSK9 result in marked elevations in LDL-C, whereas loss-of-function mutations reduce LDL-C.
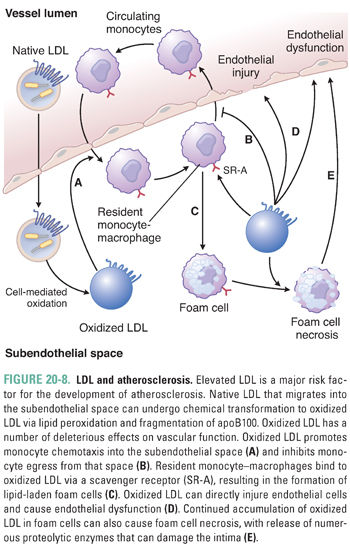
LDL particles that are not taken up by LDL receptor-expressing tissues may migrate into the intima of blood vessels and bind to proteoglycans (Fig. 20-8). There, they are subject to oxidation or nonenzymatic glycosylation. Oxidation of LDL results in lipid peroxidation and may create reactive aldehyde intermediates that fragment apoB100. The modified LDL is internalized by scavenger receptors (e.g., SR-A), which are expressed predominantly by mononuclear phagocytic cells. Unlike the LDL receptor, scavenger receptors are not down-regulated when the phagocytic cells begin to accumulate cholesterol. As a result, the continued accumulation of oxidized LDL in macrophages can lead to foam cell formation (cholesterol-rich macrophages). These foam cells may undergo apoptotic or necrotic death, releasing free radicals and proteolytic enzymes. Oxidized LDL also causes up-regulation of cytokine production, impairs endothelial function, and increases expression of endothelial adhesion molecules. All of these effects increase the local inflammatory response and promote atherosclerosis. Foam cells are a major constituent of atherosclerotic lesions, and excessive foam cell death can destabilize atherosclerotic plaques. This is attributable in part to the liberation of matrix metalloproteinases. Because plaque rupture is the main cause of acute ischemic cardiovascular events, particularly heart attacks and strokes, high plasma levels of LDL are a major risk factor for the development of atherosclerosis and subsequent cardiovascular disease. This is why Jake’s doctor became concerned when he discovered that Jake had very high plasma concentrations of LDL.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


