Ehrin J. Armstrong

In 1785, Dr. William Withering described the cardiovascular benefits of a preparation from the foxglove plant called digitalis. He used this preparation to treat patients suffering from “dropsy,” a condition in which accumulation of extravascular fluid leads to dyspnea (difficulty breathing) and peripheral edema. These symptoms are now recognized as characteristic manifestations of heart failure (HF), a clinical syndrome most commonly caused by systolic dysfunction of the left ventricle (LV). In this condition, the LV is unable to maintain adequate stroke volume despite normal filling volumes, and the LV end-diastolic volume increases in an effort to preserve stroke output. However, beyond a certain end-diastolic volume, LV diastolic pressures begin to increase, often precipitously. This increase in LV diastolic pressure results in increased left atrial and pulmonary capillary pressures, which, in turn, lead to interstitial and alveolar pulmonary edema and to increased right heart and pulmonary artery pressures. The elevated right heart pressures result in systemic venous hypertension and peripheral edema.
Dr. Withering’s use of digitalis presaged the current use of digoxin, a member of the cardiac glycoside family of drugs, to treat conditions in which myocardial contractility is impaired. Cardiac glycosides are positive inotropes, defined as agents that increase the contractile force of cardiac myocytes. Since the advent of digitalis, elucidation of the cellular mechanism of cardiac contraction has facilitated the development of other inotropic agents. After reviewing the physiology of cardiac contraction and the cellular pathophysiology of contractile dysfunction, this chapter describes four classes of positive inotropic drugs that are either approved for use or under investigation in clinical trials. An integrated discussion of therapeutic strategies for HF can be found in Chapter 26, Integrative Cardiovascular Pharmacology: Hypertension, Ischemic Heart Disease, and Heart Failure.
 GW, a 68-year-old man with known systolic dysfunction and heart failure, is admitted to the hospital with shortness of breath and nausea. GW’s cardiac history is notable for two prior myocardial infarctions, the more recent occurring about 2 years ago. Since the second infarction, he has had significant limitation of his exercise capacity. A two-dimensional echocardiogram is notable for an LV ejection fraction of 25% (normal, >55%) and moderate mitral valve regurgitation. GW has been treated with aspirin, carvedilol (a β-adrenergic receptor antagonist), captopril (an angiotensin converting enzyme inhibitor), digoxin (a cardiac glycoside), furosemide (a loop diuretic), and spironolactone (an aldosterone receptor antagonist). He has also had an automatic internal cardioverter-defibrillator (AICD) placed to prevent sustained ventricular arrhythmia and sudden cardiac death.
GW, a 68-year-old man with known systolic dysfunction and heart failure, is admitted to the hospital with shortness of breath and nausea. GW’s cardiac history is notable for two prior myocardial infarctions, the more recent occurring about 2 years ago. Since the second infarction, he has had significant limitation of his exercise capacity. A two-dimensional echocardiogram is notable for an LV ejection fraction of 25% (normal, >55%) and moderate mitral valve regurgitation. GW has been treated with aspirin, carvedilol (a β-adrenergic receptor antagonist), captopril (an angiotensin converting enzyme inhibitor), digoxin (a cardiac glycoside), furosemide (a loop diuretic), and spironolactone (an aldosterone receptor antagonist). He has also had an automatic internal cardioverter-defibrillator (AICD) placed to prevent sustained ventricular arrhythmia and sudden cardiac death.
Physical examination in the emergency department is notable for a blood pressure of 90/50 mm Hg and an irregular heart rate of 120 beats/min. An electrocardiogram indicates that the underlying cardiac rhythm is atrial fibrillation. GW is started on amiodarone (a class III antiarrhythmic), and his heart rate decreases to approximately 80 beats/min. Laboratory tests are notable for serum Na+ 148 mEq/L (normal, 135–145), BUN 56 mg/dL (normal, 7–19), K+ 2.9 mEq/L (normal, 3.5–5.1), and creatinine 4.8 mg/dL (normal, 0.6–1.2). The serum digoxin level is 3.2 ng/mL (therapeutic concentration, typically ~1 ng/mL).
Based on these findings, GW is admitted to the cardiology intensive care unit (ICU). His oral digoxin dose is held, and he is given intravenous K+ to increase his serum potassium concentration. Based on the severity of this clinical decompensation, a pulmonary artery (PA) catheter is placed to monitor cardiac pressures. GW is also started on dobutamine, and his carvedilol is held. After initiation of intravenous dobutamine, he has increased urine output and begins to feel symptomatically improved. He remains in the cardiology ICU for 7 days, and his digoxin level decreases to the therapeutic range.
Questions
1. What are the major cellular mechanisms that contribute to the pathophysiology of systolic heart failure?
2. What is the mechanism of action of digoxin?
3. What factors (including drug interactions) have contributed to digoxin toxicity in this patient?
4. Why is GW being treated with a β-adrenergic receptor antagonist and a positive inotrope (digoxin) at the same time?
5. What is the mechanism of action of dobutamine?
 PHYSIOLOGY OF CARDIAC CONTRACTION
PHYSIOLOGY OF CARDIAC CONTRACTION
The heart is responsible for receiving deoxygenated blood from the periphery, propelling this blood through the pulmonary circulation (where the hemoglobin is reoxygenated), and ultimately distributing the oxygenated blood to peripheral tissues. To accomplish the latter task, the left ventricle (LV) must develop sufficient tension to overcome the impedance to ejection that resides in the peripheral circulation. The relationship between the tension generated during the systolic phase of the cardiac cycle and the extent of LV filling during diastole is referred to as the contractile state of the myocardium. Together with preload (intraventricular blood volume), afterload (the resistance against which the left ventricle ejects), and heart rate, myocardial contractility is a primary determinant of cardiac output. Cardiac pump performance at the organ level has been studied by cardiac physiologists for many years, but now the major cellular and molecular mechanisms of cardiac contraction are understood as well.
Like skeletal muscle, cardiac muscle contracts when action potentials depolarize the plasma membranes of cardiac muscle cells. The process of excitation–contraction (EC) coupling, in which the intracellular machinery transduces an electrochemical signal into mechanical force, involves the following cascade of events: voltage-gated calcium channels open, intracellular calcium increases, contractile proteins are activated, and actin–myosin interactions shorten the contractile elements.
The cellular anatomy of ventricular myocytes is well suited to the excitation and regulation of cardiac contraction (Fig. 25-1). Specialized components of the ventricular myocyte include the sarcolemma, or myocyte plasma membrane; the sarcoplasmic reticulum (SR), a large internal membrane system that encircles the myofibrils; and the myofibrils themselves. Myofibrils are rope-like units containing precisely organized contractile proteins; the coordinated interaction of these proteins is responsible for the physical shortening of the cardiac muscle. These anatomic specializations are illustrated in Figures 25-1 and 25-2, and summarized in Table 25-1.
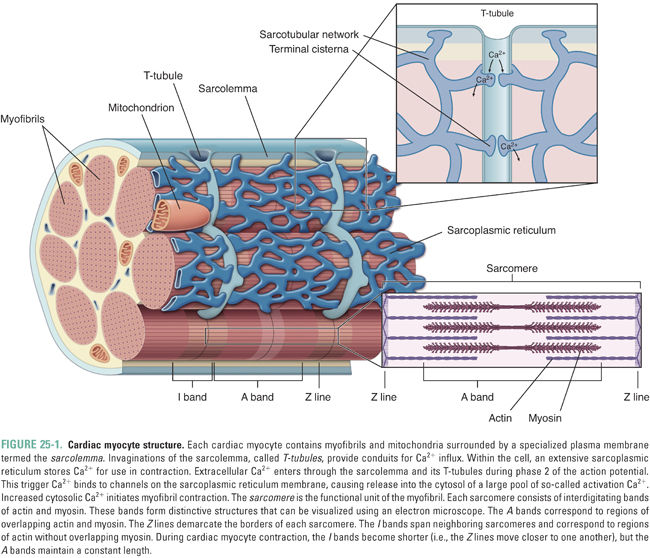
Increased cytosolic Ca2+ is the link between excitation and contraction. During the ventricular action potential (see Chapter 24, Pharmacology of Cardiac Rhythm), Ca2+ influx through L-type Ca2+ channels in the sarcolemma causes an increase in the cytosolic Ca2+ concentration. This “trigger calcium” stimulates the ryanodine receptor in the SR membrane, causing release of stored Ca2+ from the SR into the cytosol. When the Ca2+ concentration in the cytoplasm reaches approximately 10−5 M, calcium binds to troponin C and induces a conformational change in tropomyosin that releases the inhibitory protein troponin I. This release of troponin I exposes an interaction site for myosin on the actin filament, and the binding of myosin to actin initiates the contraction cycle.
Figure 25-2 illustrates the cycle by which actin–myosin interactions physically shorten the sarcomere. Each myosin filament is studded with protruding flexible heads that form reversible cross-bridges with actin filaments. Formation of actin–myosin cross-bridges, bending of the myosin heads at their flexible hinges, and detachment of the cross-bridges together allow the myosin filament to “walk up” the actin filament in both directions and thereby to pull the two ends of the sarcomere together.
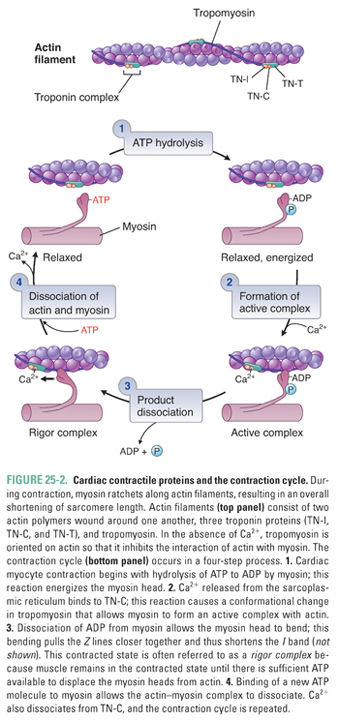
The normal function of the sarcomeric cross-bridge cycle is critically dependent on adenosine triphosphate (ATP). The ATP hydrolase (ATPase) activity of myosin provides the energy used both to drive contraction and to reset the contractile proteins, leading to relaxation. If an insufficient amount of ATP is available for cross-bridge cycling, myosin and actin remain “locked” in the associated state and the myocardium is unable to relax. This ATP dependence explains the profound impact of ischemia on both systolic contraction (the contraction cycle cannot proceed) and diastolic relaxation (actin and myosin cannot dissociate) of the myocardium.
The organization of the sarcomere and the physical mechanism of contraction explain the fundamental relationship between muscle length and tension development. Increased stretch (length) of the muscle exposes additional sites for calcium binding and for actin–myosin interaction; increased stretch also effects greater release of calcium from the SR. These cellular events provide the mechanistic explanation for the Frank-Starling law: an increase in the end-diastolic volume of the left ventricle leads to an increase in ventricular stroke volume during systole. Chapter 26 describes the organ-level implications of the Frank-Starling law in more detail.
Three major control mechanisms regulate calcium cycling and myocardial contractility in cardiac myocytes. At the sarcolemma, calcium flux is mediated by interactions between the sodium pump and sodium–calcium exchanger. At the sarcoplasmic reticulum, calcium channels and pumps regulate the extent of calcium release and reuptake. Neurohumoral influences, especially the β-adrenergic signaling pathway, further modulate calcium cycling through these channels and transporters.
The Sodium Pump and Sodium–Calcium Exchange
In the sarcolemma, three key proteins are involved in calcium regulation: the Na+/K+-ATPase, hereafter referred to as the sodium pump; the sodium–calcium exchanger; and the calcium–ATPase or calcium pump (Fig. 25-3). The activity of the sodium pump is crucial to maintain both the resting membrane potential and the concentration gradients of sodium and potassium across the sarcolemma ([Na+]out = 145 mM, [Na+]in = 15 mM, [K+]out = 5 mM, [K+]in = 150 mM). Sodium pump activity is closely linked to the intracellular calcium concentration via the sodium–calcium exchanger; this antiporter exchanges sodium and calcium in both directions across the sarcolemma. Changes in the concentration of either sodium or calcium ions inside or outside the cell affect the direction and magnitude of sodium–calcium exchange. Under normal conditions, the low intracellular sodium concentration favors sodium influx and calcium efflux. Some drugs take advantage of the functional coupling between the sodium pump and the sodium–calcium exchanger to exert their effect as positive inotropes. Digoxin, discussed in the introductory case and described in detail below, is the prototype of an inotropic agent that acts by inhibiting the sodium pump. A sarcolemmal calcium pump also helps to maintain calcium homeostasis by actively extruding calcium from the cytoplasm after cardiac contraction. A high concentration of ATP favors calcium removal (relaxation), both directly via the calcium pump and indirectly via the sodium pump.
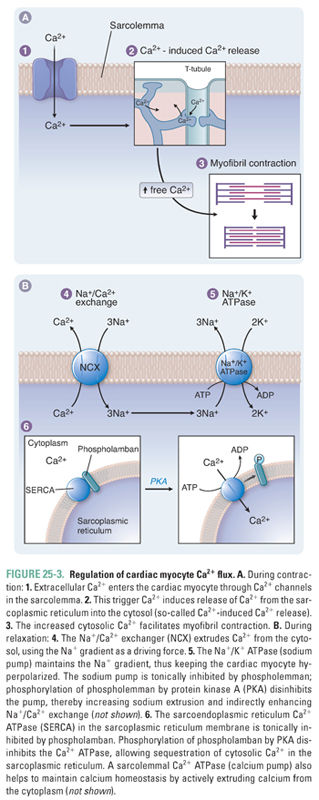
As described above, Ca2+ signaling is central to both cardiac contraction and relaxation. As such, the cardiac myocyte has well-developed systems to regulate Ca2+ flux during the cardiac cycle. In the SR, the calcium release channel (ryanodine receptor) and the calcium pump (sarcoendoplasmic reticulum calcium ATPase, SERCA) are critical to the regulation of contractility (Fig. 25-3). Proper contraction requires both that Ca2+ release into the cytoplasm is adequate to stimulate contraction and that Ca2+ reuptake into the SR is sufficient to permit relaxation and to replenish calcium stores. Cytoplasmic concentrations of both calcium and ATP regulate the activity of both the ryanodine receptor and SERCA.
As noted above, trigger calcium opens the ryanodine receptor. Cytoplasmic calcium concentration is directly related to the number of receptors that open. A safety mechanism also exists whereby high calcium levels lead to calcium–calmodulin complex formation: this complex inhibits calcium release by decreasing the open time of the ryanodine receptor. High concentrations of ATP favor the open channel conformation and thereby facilitate SR calcium release into the cytosol.
In addition to opening the ryanodine receptor, cytoplasmic calcium also stimulates SERCA, which pumps calcium back into the SR. This pump provides another control mechanism to prevent a positive feedback cycle that could irreversibly deplete the SR of calcium. As calcium pumps refill the SR, the rate of Ca2+ reuptake slows because of the declining cytoplasmic calcium concentration. ATP also favors SERCA activity; conversely, decreased ATP impairs calcium reuptake. The latter mechanism causes the rate and extent of diastolic relaxation to decrease in ischemic myocardium.
A third mediator of SERCA activity is phospholamban, an SR membrane protein that inhibits SERCA. High levels of intracellular cAMP stimulate protein kinase A to phosphorylate phospholamban, which reverses its inhibition of SERCA (Fig. 25-3). Phospholamban thus controls the rate of relaxation by regulating calcium reuptake into the SR: unphosphorylated phospholamban slows relaxation, while phosphorylated phospholamban accelerates relaxation.
Adrenergic Receptor Signaling and Calcium Cycling
β1-Adrenoceptor stimulation supports cardiac performance in several ways. First, β-receptor agonists increase β1-adrenoceptor-mediated increases in Ca2+ entry during systole; increased Ca2+ entry increases fractional shortening of cardiac muscle during contraction. This positive inotropic effect results in a higher stroke volume for any given end-diastolic volume. β-Agonists also have a positive chronotropic effect, increasing heart rate in a relatively linear dose-dependent manner. The net effect of these inotropic and chronotropic effects is to increase cardiac output:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


