David A. Barbie and David A. Frank
BIOCHEMISTRY OF INTERCELLULAR AND INTRACELLULAR SIGNAL TRANSDUCTION |

Traditional antineoplastic therapy has consisted of agents directed against DNA replication and cell division. These drugs exhibit some degree of selectivity against cancer cells, which tend to have a higher growth fraction and, in some cases, an increased susceptibility to DNA damage compared to normal cells. However, the therapeutic window of these drugs is narrow, resulting in toxicity to normal stem cells and in hematologic and gastrointestinal adverse effects. With the impressive advances in basic tumor cell biology over the last several decades and the identification of numerous oncogenes and tumor suppressor genes, the potential exists for development of agents that are targeted more specifically at the molecular circuitry responsible for the dysregulated proliferation of cancer cells. An early example of such a drug was the selective estrogen receptor modulator tamoxifen (see Chapter 30, Pharmacology of Reproduction). Tamoxifen is still one of the most active agents in the treatment of hormone receptor-positive breast cancer, with a relatively modest adverse effect profile. Subsequently, the remarkable success of imatinib mesylate in the treatment of chronic myelogenous leukemia has suggested that, in some cases, tumor cells are dependent on oncogenes such as BCR-Abl for their survival. Like tamoxifen, imatinib remains in wide use today. This chapter highlights the basic principles and agents of targeted cancer therapy, detailing recent advances and directions for the future.
 MW is a 65-year-old woman with metastatic non-small cell lung cancer. She has never smoked, and her primary tumor is an adenocarcinoma with bronchioalveolar features. Her oncologist considers treatment with carboplatin and pemetrexed but also sends genetic testing of her tumor for mutations in the epidermal growth factor receptor (EGFR) and for genetic rearrangements of anaplastic lymphoma kinase (ALK), ROS1, and RET. Her tumor is found to harbor an activating mutation in the EGFR kinase domain at codon 858, resulting in a substitution of arginine for leucine (L858R). MW is therefore treated with the oral EGFR tyrosine kinase inhibitor (TKI) erlotinib. She develops a skin rash and diarrhea but otherwise tolerates this medication well. Restaging computed tomography scans are performed 2 months after starting treatment with erlotinib. These scans reveal a dramatic reduction in MW’s tumor burden, and after 6 months, there is no residual evidence of cancer. Unfortunately, MW subsequently develops recurrence of her disease. A repeat biopsy reveals that the tumor now also shows amplification of the MET receptor tyrosine kinase. She decides to participate in a clinical trial of a MET inhibitor for treatment of her recurrent non-small cell lung cancer.
MW is a 65-year-old woman with metastatic non-small cell lung cancer. She has never smoked, and her primary tumor is an adenocarcinoma with bronchioalveolar features. Her oncologist considers treatment with carboplatin and pemetrexed but also sends genetic testing of her tumor for mutations in the epidermal growth factor receptor (EGFR) and for genetic rearrangements of anaplastic lymphoma kinase (ALK), ROS1, and RET. Her tumor is found to harbor an activating mutation in the EGFR kinase domain at codon 858, resulting in a substitution of arginine for leucine (L858R). MW is therefore treated with the oral EGFR tyrosine kinase inhibitor (TKI) erlotinib. She develops a skin rash and diarrhea but otherwise tolerates this medication well. Restaging computed tomography scans are performed 2 months after starting treatment with erlotinib. These scans reveal a dramatic reduction in MW’s tumor burden, and after 6 months, there is no residual evidence of cancer. Unfortunately, MW subsequently develops recurrence of her disease. A repeat biopsy reveals that the tumor now also shows amplification of the MET receptor tyrosine kinase. She decides to participate in a clinical trial of a MET inhibitor for treatment of her recurrent non-small cell lung cancer.
Questions
1. How does signaling through EGFR promote cell growth and survival?
2. By what mechanism does erlotinib inhibit EGFR and inhibit cancer cell growth?
3. How does amplification of MET expression lead to tumor recurrence despite treatment with erlotinib?
4. What is the most common mechanism of EGFR TKI resistance, and how might this be overcome?
 BIOCHEMISTRY OF INTERCELLULAR AND INTRACELLULAR SIGNAL TRANSDUCTION
BIOCHEMISTRY OF INTERCELLULAR AND INTRACELLULAR SIGNAL TRANSDUCTION
Growth Factors and Growth Factor Receptors
External signals stimulate cell growth and proliferation via the interaction of growth factors with specific cell surface receptors. Growth factor receptors typically contain an extracellular ligand-binding domain, a hydrophobic transmembrane domain, and a cytoplasmic domain that has either intrinsic tyrosine kinase activity or an associated protein tyrosine kinase (Fig. 40-1). In general, binding of the growth factor ligand results in receptor oligomerization, a conformational change in the cytoplasmic domain of the receptor, and tyrosine kinase activation. Intracellular targets are subsequently phosphorylated, propagating a signal that culminates in progression through the cell cycle and cellular proliferation.
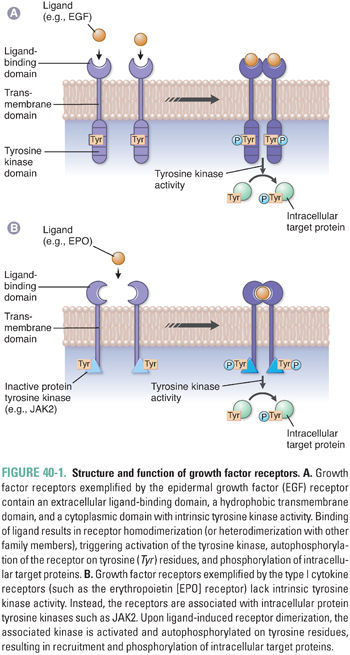
One example of a receptor tyrosine kinase is the epidermal growth factor receptor (EGFR), which possesses intrinsic tyrosine kinase activity and is a member of the broader ErbB family of proteins, including EGFR (ErbB1), HER2/neu (ErbB2), ErbB3, and ErbB4. Binding of epidermal growth factor (EGF) or transforming growth factor-α (TGF-α) to EGFR results in receptor homodimerization and propagation of a growth signal. In addition, heterodimerization between family members can occur, yielding further diversity in the signal that is transduced. ErbB receptors are expressed on epithelial cells and are often activated or overexpressed in a variety of carcinomas (e.g., EGFR in non-small cell lung cancer and HER2/neu in breast cancer).
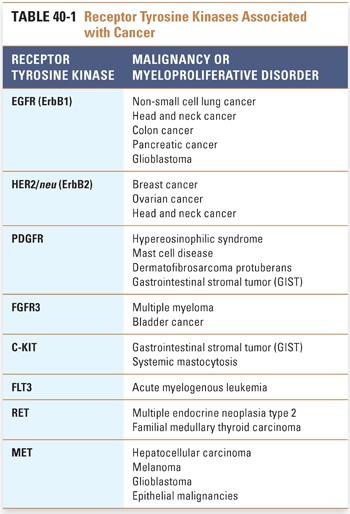
Other examples of receptor tyrosine kinases include the insulin-like growth factor receptor 1 (IGF1R), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), C-KIT, Bruton’s tyrosine kinase (BTK), FMS-like tyrosine kinase (FLT3), anaplastic lymphoma kinase (ALK), ROS1, RET, and MET. Signaling through these receptors activates the growth of certain hematopoietic and mesenchymal tissues, and dysregulation of these receptors is frequently observed in specific myeloproliferative disorders, leukemias, sarcomas, and epithelial cancers (Table 40-1).
Other hematopoietic receptors rely on interaction with an associated cytoplasmic tyrosine kinase for transduction of a growth signal. For example, type I cytokine receptors such as the erythropoietin receptor (EpoR), thrombopoietin receptor (TpoR), and G-CSF receptor (GCSFR) form specifically oriented homodimers upon binding of ligand, resulting in activation of the associated tyrosine kinase JAK2, which leads to further signaling and ultimately to cell growth. Activating mutations of the receptors themselves (e.g., EpoR) have been implicated in conditions such as congenital polycythemia. A common activating mutation in JAK2, resulting in the conversion of valine to phenylalanine at position 617 (V617F), has been found in a majority of patients with the myeloproliferative disorder polycythemia vera and in a significant fraction of patients with essential thrombocythemia and myeloid metaplasia with myelofibrosis.
Intracellular Signal Transduction Pathways
Activation of a growth factor receptor initiates the transduction of a series of intracellular signals, culminating in events such as cell cycle entry, promotion of protein translation and cell growth, and enhanced cell survival. Two broad categories of pathways activated by receptor tyrosine kinases are the RAS-MAP kinase pathway and the phosphatidylinositol-3-kinase (PI3K)-AKT pathway (Fig. 40-2).
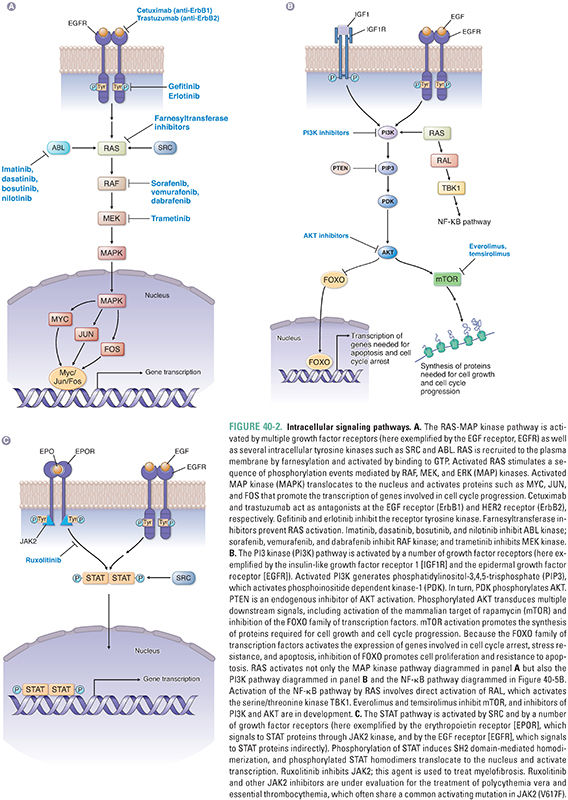
The Kirsten ras gene was initially identified as a retroviral oncogene in rats and subsequently found to have several human homologues, including K-ras, H-ras, and N-ras. The protein (RAS) encoded by ras is targeted to the plasma membrane by the farnesyltransferase-mediated addition of a hydrophobic farnesyl group to its COOH-terminus; this targeting brings RAS into close proximity to activated receptor tyrosine kinases. Intracellular nonreceptor tyrosine kinases such as ABL and SRC, also originally identified as oncogene products, can activate signaling through RAS as well (Fig. 40-2A).
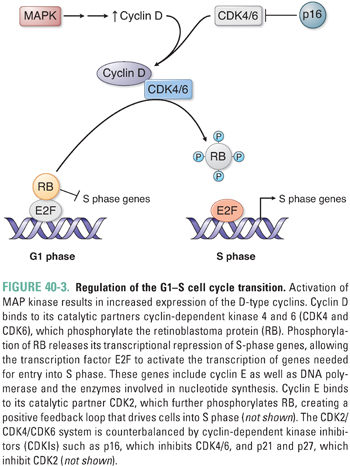
Upon activation by binding to GTP, RAS triggers a series of phosphorylation events mediated by the kinases RAF, MEK, and ERK (MAP kinase), the targets of which include transcription factors that promote activation of genes involved in proliferation. For example, activation of cyclin D transcription results in cyclin D expression and binding to its catalytic partners, cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) (Fig. 40-3). These complexes initiate phosphorylation of the retinoblastoma protein (pRB), thereby lifting pRB’s repression of the transcription factor E2F. E2F mediates the expression of components of the DNA replication machinery and enzymes involved in nucleotide synthesis. Thus, phosphorylation of pRB by cyclin D–CDK4/6 and subsequent activation of other cyclin–CDK complexes (such as cyclin E–CDK2) result in the transition from G1 to S phase and progression through the cell cycle. While such signaling cascades might seem unnecessarily complicated, they allow for the integration of diverse extracellular and intracellular signals, the opportunity for multiple points of feedback control, and the tight regulation of critical events such as cellular proliferation.
A second key intracellular signaling pathway is controlled by the lipid kinase PI3K. Stimulation of receptors for growth factors such as insulin or insulin-like growth factors (IGFs) commonly leads to activation of PI3K via an associated insulin receptor substrate protein (IRS). ErbB family members can also activate this pathway via phospholipase C-γ (PLC-γ), and RAS can also promote signaling through this pathway (Fig. 40-2B). Activation of PI3K results in generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from plasma membrane phospholipids, activation of phosphoinositide dependent kinase-1 (PDK-1) via translocation to the cell membrane, and phosphorylation of AKT by PDK-1. This pathway is negatively regulated by the lipid phosphatase PTEN, which degrades PIP3. Downstream effects of AKT activation include promotion of translation and cell growth by the mammalian target of rapamycin (mTOR). In addition, phosphorylation of the forkhead family of transcription factors (FOXO) by AKT results in their exclusion from the nucleus, preventing expression of genes involved in cell cycle arrest, stress resistance, and apoptosis. Thus, a net effect of activating the PI3K-AKT pathway is the promotion of cell survival. Activating mutations in the catalytic subunit of PI3K (PI3KCA) and inactivating mutations in PTEN are frequently observed in a variety of malignancies, including breast cancer, colon cancer, prostate cancer, and glioblastoma.
In addition to activating the MAP kinase and PI3K pathways, RAS also activates the RAL signaling pathway (Fig. 40-2B). RAL signaling remains incompletely understood, but this pathway activates specific kinases involved in innate immunity and vesicle trafficking. RAL signaling is also essential for RAS-mediated tumorigenesis.
Signaling via type I cytokine receptors is associated with activation of the JAK-STAT pathway (Fig. 40-2C). Receptor dimerization activates JAKs, or Janus kinases, via transphosphorylation, allowing the recruitment of STAT proteins through their SH2 domains. STATs are a family of proteins that shuttle from the cytoplasm to the nucleus to regulate transcription directly. Phosphorylation of STAT proteins by JAKs leads to the formation of SH2 domain-mediated STAT homo- or heterodimers that translocate to the nucleus and regulate transcription. Growth factor receptors such as EGFR, as well as intracellular tyrosine kinases such as SRC, can also signal through activation of STATs.
Proteasome Structure and Function
Key cellular processes such as cell cycle progression and apoptosis are also regulated at the post-translational level by protein degradation. One of the major systems involved in this control is the ubiquitin–proteasome pathway, which comprises three enzymes that target specific proteins for ubiquitin conjugation and degradation by the proteasome (Fig. 40-4A). Ubiquitin is a 9-kDa protein that derives its name from its widespread distribution in tissues and its conservation across eukaryotes. The first enzyme involved in the process, E1, uses ATP to activate ubiquitin. The second enzyme in the cascade, E2, is a ubiquitin-conjugating enzyme that transiently carries ubiquitin and acts in conjunction with the third enzyme, the ubiquitin ligase E3, to form a polyubiquitin chain that is transferred to the target protein on an internal lysine residue.
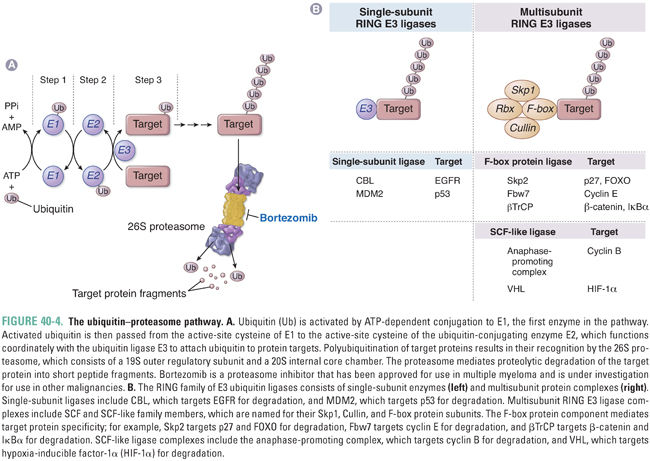
E1 is nonspecific, and there are a number of different E2 ubiquitin-conjugating enzymes with a limited degree of specificity. The E3 ubiquitin ligase component is largely responsible for target protein specificity. The RING family of E3 ligases contains a characteristic RING finger domain with conserved histidine and cysteine residues complexed with two central Zn2+ ions. RING E3 ligases can be subdivided into single-subunit E3 ligases and multisubunit complexes such as the Skp1-Cullin-F-box protein family (SCF) E3 ligases. In the latter complexes, the RING finger component, Rbx, is distinct from the specificity component, the F-box protein, which is so named because of a characteristic motif first identified in cyclin F.
Once proteins are selectively ubiquitinated, they are targeted for degradation by the 26S proteasome, which is a cylindrical particle present in both cytoplasm and nucleus. The core 20S subunit is the catalytic component with multiple proteolytic sites, while the 19S regulatory component mediates binding to ubiquitin-conjugated proteins and has multiple ATPases involved in substrate unfolding and delivery to the central 20S chamber. Substrates are cleaved progressively, with one protein being completely degraded before the next protein enters. Short peptide segments, on average 6 to 10 amino acids in length, are extruded and subsequently hydrolyzed to individual amino acids in the cytosol.
Regulation of protein degradation occurs largely at the level of the E3 ubiquitin ligase and governs key aspects of cell cycle control, apoptosis, and other important cellular processes (Fig. 40-4B). For example, CBL is a single-subunit RING E3 ubiquitin ligase that targets phosphorylated EGFR family members for degradation. In addition, both cyclins and cyclin-dependent kinase inhibitors are major targets for ubiquitin-mediated proteasomal degradation. The anaphase-promoting complex is a multisubunit RING-containing E3 ligase that is activated by phosphorylation late in mitosis, triggering degradation of cyclin B and progression through mitosis. Regulation of the G1–S cell cycle transition is in part mediated by the cyclin-dependent kinase inhibitor p27, which inhibits cyclin E–CDK2 and cyclin A–CDK2 complexes. Degradation of p27 is regulated by another SCF E3 ligase, which binds p27 via its F-box specificity component Skp2. Thus, overexpression of Skp2, which is found in a number of tumor types, can promote cell cycle progression by degrading p27. Degradation of FOXO by Skp2 is a second mechanism by which overexpression of Skp2 may promote tumorigenesis. Yet another SCF E3 ligase complex regulates cyclin E activity by targeting it for degradation via the F-box protein Fbw7. Loss of Fbw7 has been implicated in tumor progression due to high levels of cyclin E.
Another example of an E3 ligase with a critical role in the regulation of apoptosis and cell cycle regulation is MDM2, a single-subunit RING E3 ligase that targets p53 for degradation. Activation of MDM2 is linked to impairment of apoptosis and promotion of tumorigenesis via loss of p53. MDM2 is inhibited by the p14ARF protein, which shares the same genomic locus as the CDK4/6 inhibitor p16. Disruption of this locus, which is one of the most common events in cancer, leads ultimately to both p53 and pRB inactivation.
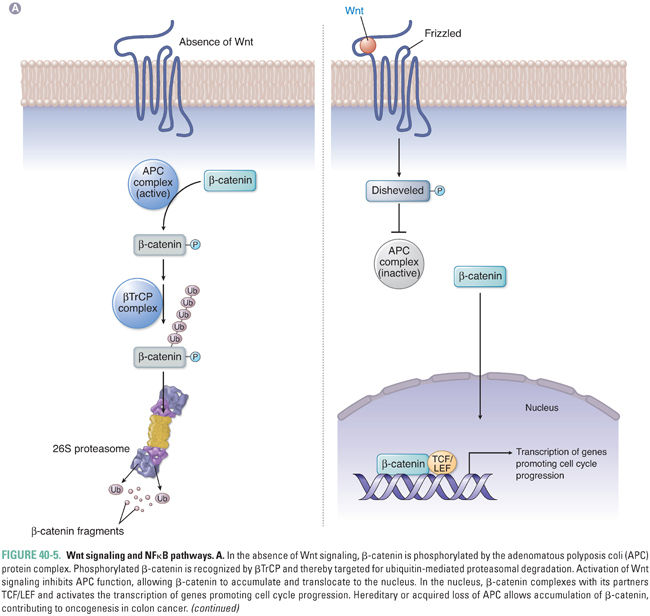
Other key cellular pathways regulated by ubiquitin-mediated proteasomal degradation include the Wnt signaling pathway and the nuclear factor-kappa B (NFκB) pathway. Both pathways are targeted by the common F-box protein βTrCP, which recognizes phosphorylated substrates (Fig. 40-5). Activation of Wnt signaling prevents phosphorylation of β-catenin, which allows it to escape recognition by βTrCP and ubiquitin ligation by SCF E3 ligase. Unphosphorylated β-catenin then translocates to the nucleus with its partners TCF/LEF and activates transcription of genes such as myc and cyclin D1. This pathway is also regulated by the adenomatous polyposis coli (APC) gene, which forms part of the complex that promotes phosphorylation and subsequent destruction of β-catenin. Loss of APC in colorectal cells prevents phosphorylation of β-catenin, leading to β-catenin accumulation and to promotion of cancer.
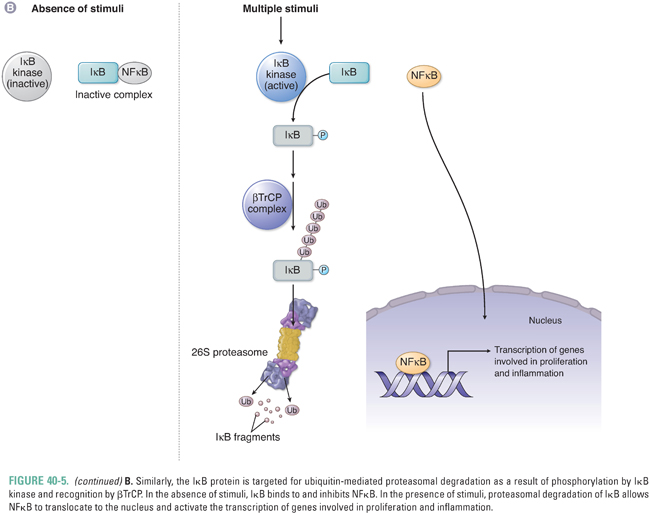
The F-box protein βTrCP also regulates signaling through NFκB, which is inhibited by its association with the inhibitor of NFκB (IκB). Phosphorylation of IκB by a family of IκB kinases (IKKs) allows βTrCP to bind to IκB and activate its proteasome-mediated degradation. The release of IκB inhibition allows NFκB to translocate to the nucleus and activate transcription of genes involved in inflammation, proliferation, and survival. Specific IKKs may be aberrantly activated in cancer cells and thereby generate an environment favoring tumor cell survival.
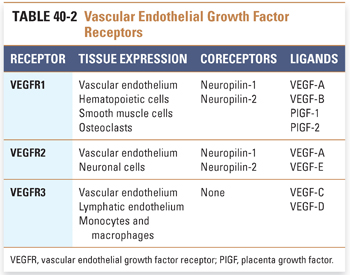
Solid tumors require development of a neovasculature in order to sustain growth and survive conditions of hypoxia. Tumor angiogenesis is a complex process involving a number of different pro- and antiangiogenic factors. The vascular endothelial growth factor (VEGF) family of proteins and receptors has emerged as a key regulator of this process. The VEGF family consists of seven ligands, including VEGF-A, -B, -C, -D, and -E and placenta growth factor (PlGF)-1 and -2 (Table 40-2). These ligands have varying affinities for the major VEGF receptors, VEGFR1 (also known as Flt-1), VEGFR2 (Flk-1/KDR), and VEGFR3 (Flt-4). The VEGF receptors are receptor tyrosine kinases. Neuropilins (NRP-1 and -2) are coreceptors that lack an intracellular signaling domain and enhance the binding of ligand to VEGFR1 and VEGFR2. VEGFR1 and VEGFR2 are expressed on the vascular endothelium and play key roles in angiogenic signaling, while signaling through VEGFR3 appears to play a major role in lymphangiogenesis (i.e., development of new lymphatic vessels). VEGFR2, which appears to be the major proangiogenic receptor targeted by VEGF-A, signals via both a RAF/MAP kinase pathway to promote proliferation of endothelial cells and a PI3K/AKT pathway to promote endothelial cell survival. VEGF also potently induces vascular permeability, utilizing similar signaling pathways both to promote the formation of transendothelial vesicular organelles and to open interendothelial junctions. Invasion and migration of endothelial cells is promoted by activation of matrix metalloproteinases and serine proteases and by reorganization of intracellular actin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


