Robert S. Griffin and Clifford J. Woolf
Sensory Transduction: Excitation of Primary Afferent Neurons |
Descending and Local Inhibitory Regulation in the Spinal Cord |
Nonsteroidal Anti-Inflammatory Drugs and Nonopioid Analgesics |

Everyone has experienced pain in response to an intense or noxious stimulus. This physiologic “ouch” pain helps us to avoid potential damage by acting as an early warning or protective signal. Pain can, however, also be incapacitating, as after trauma, during recovery from surgery, or in association with medical conditions that are characterized by inflammation, such as rheumatoid arthritis. Under circumstances where tissue injury and inflammation are present, noxious stimuli elicit more severe pain than normal because of increases in the excitability of the somatosensory system, and stimuli that would not normally cause pain become painful. In addition, nerve injury produced by disease or trauma—as in amputation, HIV infection, varicella-zoster (VZV) infection, cytotoxic treatment, and diabetes mellitus—evokes pain that persists long after the initiating cause has disappeared. In these conditions, pathologic and sometimes irreversible alterations in the structure and function of the nervous system lead to severe and intractable pain. For such patients, the pain is the pathology rather than a physiologic defense mechanism. Finally, there are patients who experience considerable pain in the absence of noxious stimuli, inflammation, or lesions to the nervous system. This dysfunctional pain, as in tension-type headache, fibromyalgia, or irritable bowel syndrome, results from an abnormal function of the nervous system.
These categories of pain—physiologic, inflammatory, neuropathic, and dysfunctional—are produced by different mechanisms. Ideally, treatment should be targeted at the specific mechanisms that produce pain rather than at suppressing the symptom of pain. That said, many of the currently available pharmacologic agents relieve pain by suppressing the symptom. The mechanisms of action of drugs that relieve pain involve interference with the response of primary sensory neurons to somatic or visceral sensory stimuli, inhibition of the relaying of pain information to the brain, and blockade of the perceptual response to a painful stimulus. In this chapter, the discussion of pain and analgesic pharmacology begins by describing the mechanisms by which noxious stimuli lead to the perception of pain. The chapter continues by considering the processes responsible for the heightened pain sensitivity that occurs in response to inflammation and lesions of the nervous system. The discussion concludes by describing the mechanisms of action of the major drug classes used for clinical pain relief.
 JD, a 15-year-old boy, is severely burned while escaping from a building fire. The extensive burns include first- and second-degree burns covering much of his body and a local, full-thickness burn on his right forearm. He reaches the emergency department in severe pain and is treated with intravenous morphine in increasing quantities until he reports that the pain has subsided. This dose of morphine is then maintained. The next day, he has surgical debridement of his burn wounds and a skin graft to his right forearm. During the operation, an anesthesiologist provides a continuous intravenous infusion of remifentanil, with a bolus dose of morphine added near the end of the operation. At the end of the operation, and for 4 days thereafter, JD receives intravenous morphine through a patient-controlled analgesia device. As the burns heal, the morphine dose is tapered and eventually replaced with an oral oxycodone/acetaminophen combination tablet. Three months later, JD reports severe loss of sensation to touch in the area of the skin graft. He also describes a persistent tingling sensation in this area, with occasional bursts of sharp, knife-like pain. After referral to a pain clinic, JD is prescribed oral gabapentin, which partially reduces his symptoms. However, he reports to the pain clinic again 2 months later, still in severe pain. At this time, amitriptyline is added to the gabapentin, and the pain is further relieved. Three years later, JD’s lingering pain has resolved and he no longer requires medication, but the lack of forearm sensation persists.
JD, a 15-year-old boy, is severely burned while escaping from a building fire. The extensive burns include first- and second-degree burns covering much of his body and a local, full-thickness burn on his right forearm. He reaches the emergency department in severe pain and is treated with intravenous morphine in increasing quantities until he reports that the pain has subsided. This dose of morphine is then maintained. The next day, he has surgical debridement of his burn wounds and a skin graft to his right forearm. During the operation, an anesthesiologist provides a continuous intravenous infusion of remifentanil, with a bolus dose of morphine added near the end of the operation. At the end of the operation, and for 4 days thereafter, JD receives intravenous morphine through a patient-controlled analgesia device. As the burns heal, the morphine dose is tapered and eventually replaced with an oral oxycodone/acetaminophen combination tablet. Three months later, JD reports severe loss of sensation to touch in the area of the skin graft. He also describes a persistent tingling sensation in this area, with occasional bursts of sharp, knife-like pain. After referral to a pain clinic, JD is prescribed oral gabapentin, which partially reduces his symptoms. However, he reports to the pain clinic again 2 months later, still in severe pain. At this time, amitriptyline is added to the gabapentin, and the pain is further relieved. Three years later, JD’s lingering pain has resolved and he no longer requires medication, but the lack of forearm sensation persists.
Questions
1. What mechanisms produced and sustained the pain that lasted from JD’s exposure to the fire until his initial treatment?
2. What was the rationale for the sequence of medications used during the skin debridement operation?
3. Explain the mechanisms that could produce spontaneous pain in the region of the full-thickness burn months to years after healing of the skin and the rationale for using gabapentin to treat JD’s chronic pain.
4. Why was morphine tapered gradually and replaced with a combination oxycodone/acetaminophen tablet?

Pain is the end perceptual consequence of the neural processing of particular sensory information. The initial stimulus usually arises in the periphery and is transferred under multiple controls through sensory relays in the central nervous system (CNS) to the cortex. This system can be usefully analyzed in terms of the sites of action at which drugs intervene to produce analgesia. First, transduction of intense external, noxious stimuli depolarizes the peripheral terminals of “high-threshold” primary sensory neurons. The primary sensory neurons, called nociceptors because they respond to noxious stimuli, are high-threshold because they require a strong, potentially tissue-damaging stimulus to depolarize their terminals. The resulting action potentials are conducted to the CNS by the axons of the primary afferent sensory neurons, running first in peripheral nerves and then in dorsal roots, which then synapse on neurons in the dorsal horn of the spinal cord. The secondary projection neurons transmit information to the brainstem and thalamus, which then relay signals to the cortex, hypothalamus, and limbic system. Transmission is modulated at all levels of the nervous system by remote and local circuit inhibitory and excitatory interneurons (Fig. 18-1).
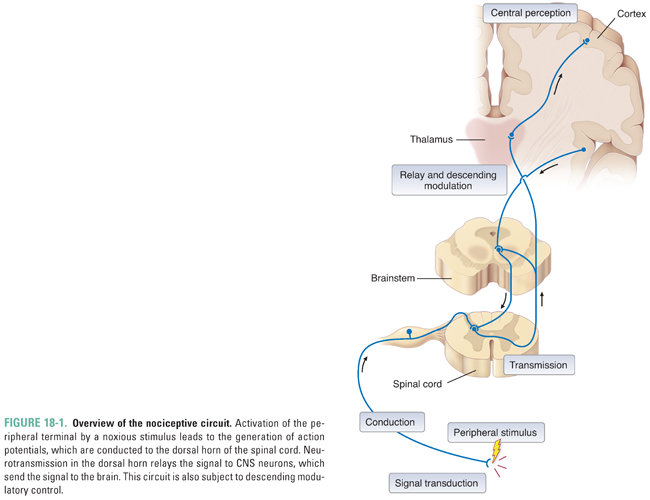
Sensory Transduction: Excitation of Primary Afferent Neurons
The peripheral terminals of primary afferent somatic and visceral sensory nociceptor fibers respond to thermal, mechanical, and chemical stimuli (Fig. 18-2). Highly specialized ion channels/receptors undergo conformational changes in response to one or more of these stimuli and thereby mediate the depolarization (generator potential) necessary to initiate an action potential. The frequency and duration of the action potentials in the activated fiber transfer information to the CNS about the onset, intensity, and duration of the stimulus, while the spatial localization of the fiber’s central terminal in the dorsal horn of the spinal cord encodes the site of the stimulus.
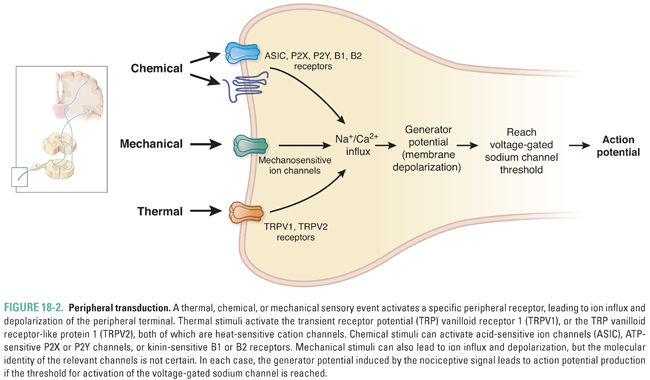
Thermal pain sensitivity depends on distinct populations of primary sensory neurons: some become active at cold temperatures (<16°C), whereas others respond to heat. Heat pain-sensing neurons produce action potentials at temperatures higher than 42°C. Responses to noxious heat involve thermosensitive nonselective cation channels, particularly TRPV1, which is a member of the transient receptor potential (TRP) family of ion channels. This channel becomes active in response to low extracellular pH, vanilloid chemical ligands such as capsaicin (the pungent ingredient in chili peppers), or heat in excess of 42°C. In addition to TRPV1, other TRP channels with varying activation temperatures are involved. TRPV heat-sensitive ion channels represent targets for the development of new drugs to interfere with peripheral heat sensation, although this may also interfere with body temperature regulation. In JD’s case, the initial experience of pain was mediated by heat activation of thermosensitive high-threshold peripheral neurons expressing TRPV1. Cool is detected by the TRP channel TRPM8 and intense cold may be detected by TRPA1. TRPM8 is also activated by menthol and TRPA1 by allyl isothiocyanate, the pungent ingredient in mustard and wasabi.
Similarly, a specific subpopulation of primary afferent terminals (the high-threshold mechanonociceptors) is excited by relatively intense mechanical stimuli, such as a pinch or a pinprick. The mechanonociceptor for innocuous tactile stimulation is a very large channel called piezo 2, while the transducer for noxious mechanotransduction has not yet been identified.
The peripheral terminals of nociceptor neurons respond not only to thermal and mechanical stimuli but also to multiple chemical signals. Some chemical agents directly excite peripheral terminals (chemical activators), whereas others increase the sensitivity of the peripheral terminals (sensitizing agents). Most known chemical ligands that evoke a somatosensory response are associated with cell injury or inflammation. These chemical ligands include protons, potassium ions, ATP, amines, prostanoids, cytokines, chemokines, nerve growth factor, and bradykinin. For example, cardiac angina is a nociceptive event that involves activation of visceral chemotransducers in nociceptor neurons innervating the heart. These chemotransducers are activated by protons that are released by inadequately perfused myocardial tissue.
Several different types of chemical stimuli can excite nociceptor neurons (Table 18-1). Low extracellular pH, which occurs in ischemia and inflammation, produces depolarizing cation influx through TRPV1 and likely also via acid-sensitive ion channels (ASICs). Elevated extracellular ATP concentration also signals cell injury, because cell rupture releases millimolar concentrations of ATP into the extracellular space (where the ATP concentration is normally very low). Two major classes of ATP receptor include the P2X ligand-gated channels and the P2Y G protein-coupled ATP receptors.
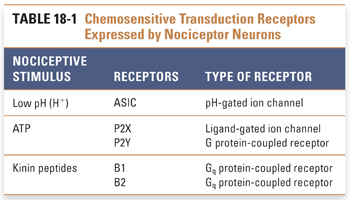
Kinins are a third set of chemical stimuli that excite the peripheral terminals of sensory neurons. Kinin peptides are produced from kininogens by the action of kallikrein serine proteases; this process usually occurs in the setting of inflammation and tissue damage. Kinins act by stimulating bradykinin B1 and B2 receptors. The B2 receptor is constitutively expressed throughout the nervous system, while expression of the B1 receptor is induced in response to bacterial lipopolysaccharide, inflammatory cytokines, and peripheral nerve injury. Both kinin receptors are G protein-coupled and increase intracellular calcium by production of inositol 1,4,5-trisphosphate. Activation of the B2 receptor also leads to the formation of prostaglandins E2 and I2. In the introductory case, as the heat sensation was followed by burn injury, these chemical mediators likely further contributed to JD’s pain. Bacterial pathogens can also directly activate nociceptors via formylated peptides acting on G protein-coupled receptors and via secretion of toxins such as alpha hemolysin, which is a channel-like protein that binds to certain nociceptors and thereby contributes to the pain of bacterial infection.
Conduction from the Periphery to theSpinal Cord
The axons of primary afferent neurons conduct information from the peripheral terminal to the CNS. These neurons can be classified into three major groups according to their conduction velocity and caliber; these groups also have distinct stimulus sensitivities and distinct central termination patterns. The first group (Aβ) consists of rapidly conducting fibers that respond with a low stimulus threshold to mechanical stimuli and are activated by light touch, vibration, or movement of hairs. Aβ-fibers synapse on CNS neurons located in the dorsal horn of the spinal cord and in dorsal column nuclei of the brainstem. The second population (Aδ) includes fibers that conduct with intermediate velocity and respond to cold, heat, or low- or high-intensity mechanical stimuli. The third group (C-fibers) conduct slowly, synapse in the spinal cord, and typically respond multimodally; they are capable of producing action potentials in response to heat, warmth, intense and innocuous mechanical stimuli, or chemical irritants (polymodal nociceptors, tactile detectors, and itch-provoking pruriceptors). Some C-fiber afferents (referred to as silent or sleeping nociceptor fibers) cannot be activated normally but become responsive only during inflammation. Aδ- and C-fibers terminate in the most superficial laminae of the dorsal horn (lamina I and II).
For conduction to occur, voltage-gated sodium channels must convert depolarization of the peripheral terminal into an action potential. Six types of voltage-gated sodium channels are expressed in primary afferent neurons, of which three, Nav1.7, Nav1.8, and Nav1.9, are expressed uniquely in primary afferents. Gain-of-function mutations in Nav1.7 produce hyperexcitability of nociceptors and thereby contribute to primary erythromelalgia, an inherited condition associated with severe burning pain that is either spontaneous or provoked in response to mild thermal stimuli. Loss-of-function mutations in Nav1.7 result in congenital insensitivity to pain, highlighting the critical role of this channel in nociception and its potential attractiveness as a target for analgesics. Nav1.8 and Nav1.9 are selectively expressed in small-caliber neurons, most of which respond only to high-threshold peripheral stimuli (nociceptors). These two channel types also have higher activation thresholds and inactivate more slowly than other neuronal voltage-gated sodium channels. Because of their specific expression pattern in pain fibers, selective sodium channel blockers represent future pharmacologic targets of particular interest, especially if they produce a use-dependent block.
Currently, the topical or regional use of nonselective, sodium channel-blocking local anesthetic agents is a mainstay for the treatment of acute postoperative and procedural pain (see Chapter 12, Local Anesthetic Pharmacology). Sodium channel-blocking antiepileptic and antiarrhythmic drugs (see Chapter 16, Pharmacology of Abnormal Electrical Neurotransmission in the Central Nervous System, and Chapter 24, Pharmacology of Cardiac Rhythm, respectively) are also used for certain neuropathic pain conditions, particularly trigeminal neuralgia.
Transmission in the Dorsal Horn of theSpinal Cord
Action potentials generated in primary afferents induce neurotransmitter release upon reaching their central axon terminals in the dorsal horn of the spinal cord. N-type voltage-gated calcium channels have a substantial role in controlling neurotransmitter release from synaptic vesicles. Gabapentin and pregabalin are antiepileptic drugs that act on the alpha 2 delta calcium channel subunit. Via a mechanism that is not completely understood but involves disruption of calcium channel trafficking to the membrane, these agents likely modulate CNS transmission of nociceptive information. Although their effectiveness is limited, both agents are widely used in the treatment of chronic neuropathic pain because of their generally favorable adverse effect profile. A naturally occurring snail poison, omega-conotoxin, acts as a selective N-type calcium channel blocker; a synthetic mimic of this peptide, ziconotide, is currently used to treat severe pain conditions. However, such calcium channel blockers also alter the function of sympathetic neurons (producing hypotension) and many central neurons (affecting cognitive function). Thus, the use of ziconotide is limited to the highly specialized scenario of intrathecal administration because it is necessary to limit the drug’s effects to the spinal cord.
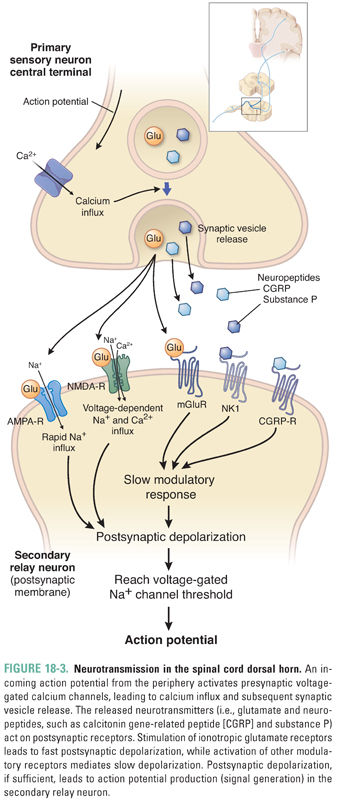
Synaptic transmission takes place between C-fiber primary afferents and secondary projection neurons in the dorsal horn. This transmission has fast and slow components (Fig. 18-3). Acting on ionotropic AMPA and NMDA receptors, glutamate mediates fast excitatory transmission between primary and secondary sensory neurons. Acting on metabotropic mGluR receptors, glutamate also mediates a slow synaptic modulatory response. Neuropeptides, such as the tachykinins substance P and calcitonin gene-related peptide (CGRP), as well as other synaptic neuromodulators, including the neurotrophin brain-derived neurotrophic factor (BDNF), are co-released with glutamate and also produce slow synaptic effects by acting on metabotropic G protein-coupled receptors and receptor tyrosine kinases. The presence of these co-released peptides allows considerable use-dependent functional plasticity of pain transmission. The physiologic function of the neuropeptides in synaptic transmission involves signaling responses to stimuli of particularly high intensity, because release of neuropeptide-containing synaptic vesicles requires higher frequency and longer lasting action potential trains than release of glutamate-containing vesicles. New strategies to target CGRP and its actions are being developed, particularly for migraine.
Descending and Local Inhibitory Regulation in the Spinal Cord
Synaptic transmission in the spinal cord is regulated by the actions of both local inhibitory interneurons and projections that descend from the brainstem to the dorsal horn. Because these systems can limit transfer of incoming sensory information to the brain, they represent an important site for pharmacologic intervention. The major inhibitory neurotransmitters in the dorsal horn of the spinal cord are opioid peptides, norepinephrine, serotonin (5-HT), glycine, and GABA (Fig. 18-4). The physiology of GABA receptors is discussed in Chapter 13, Pharmacology of GABAergic and Glutamatergic Neurotransmission.
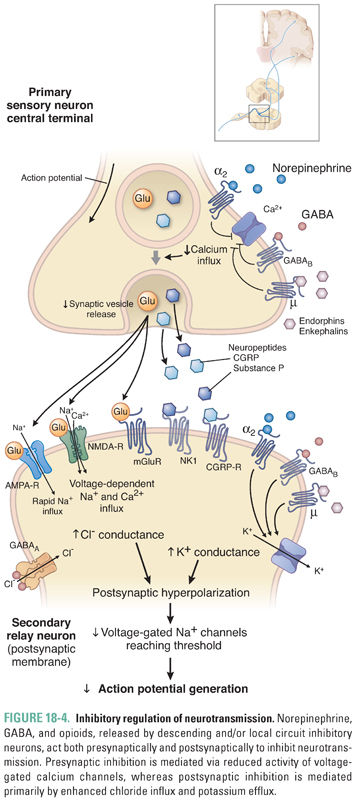
The opioid peptides inhibit synaptic transmission and are released at several CNS sites in response to noxious stimuli. All endogenous opioid peptides, which include β-endorphin, the enkephalins, and the dynorphins, share the N-terminal sequence Tyr-Gly-Gly-Phe-Met/Leu. The opioids are proteolytically released from the larger precursor proteins pro-opiomelanocortin, proenkephalin, and prodynorphin. Opioid receptors fall into three classes, designated μ, δ, and κ, all of which are seven-transmembrane G protein-coupled receptors. The μ-opioid receptors mediate morphine-induced analgesia. This conclusion is based on the observation that the μ-opioid receptor knockout mouse exhibits neither analgesia nor adverse effects in response to morphine administration. The endogenous opioid peptides are receptor-selective: the dynorphins act primarily on κ receptors, while both enkephalins and β-endorphin act on μ and δ receptors. The physiologic role of the endogenous opioid peptides remains poorly understood, although they may mediate reward effects—for example, after sunburn. The effects of opioid receptor signaling include reduced presynaptic calcium conductance, enhanced postsynaptic potassium conductance, and reduced adenylyl cyclase activity. The first function impedes presynaptic neurotransmitter release; the second reduces postsynaptic neuronal responses to excitatory neurotransmitters; the physiologic role of the third remains unknown.
Opioids produce analgesia because of their action in the brain, brainstem, spinal cord, and peripheral terminals of primary afferent neurons. In the brain, opioids alter mood, produce sedation, and reduce the emotional reaction to pain. In the brainstem, opioids increase the activity of cells that provide descending inhibitory innervation to the spinal cord; here, opioids also produce nausea and respiratory depression. Spinal opioids inhibit synaptic vesicle release from primary afferents and hyperpolarize postsynaptic neurons (see above). Evidence also exists that peripheral opioid receptor stimulation reduces the activation of primary afferents and modulates immune cell activity. Action of opioids at these serially located sites is thought to have a synergistic effect to inhibit information flow from the periphery to the brain.
Norepinephrine is released by projections that descend from the brainstem to the spinal cord. The α2-adrenergic receptor, a seven-transmembrane G protein-coupled receptor (see Chapter 11, Adrenergic Pharmacology), is the primary receptor for norepinephrine in the spinal cord. As with opioid receptor activation, α2-adrenergic receptor activation inhibits presynaptic voltage-gated calcium channels, opens postsynaptic potassium channels, and inhibits adenylyl cyclase. Because α2-adrenergic receptors are expressed both presynaptically and postsynaptically, spinal norepinephrine release can both reduce presynaptic vesicle release and decrease postsynaptic excitation. The α2-adrenergic receptor agonist clonidine is sometimes used to treat pain, although this application is limited by adverse effects that include sedation and postural hypotension. Serotonin is also released in the spinal cord by projections descending from the brainstem. This neurotransmitter acts on several receptor subtypes that mediate both excitatory and inhibitory effects on nociception. The 5-HT3 ligand-gated channel may be responsible for the excitatory actions of serotonin in the spinal cord; several of the 5-HT G protein-coupled receptors may mediate the inhibitory actions of 5-HT. Given this complexity, the mechanism of the analgesic effect of serotonin is not fully understood. Selective serotonin reuptake inhibitors have been tested in the treatment of pain but have generally had little beneficial effect. Selective norepinephrine (NE) reuptake inhibitors do have analgesic action, as do dual NE/5-HT reuptake inhibitors such as duloxetine—this agent is used to treat many chronic pain conditions. Tramadol, a weak centrally acting opioid, also has monoaminergic actions and is widely used to treat mild pain. Its relatively weak efficacy as a single agent is increased when combined with acetaminophen, and its lack of abuse potential makes the drug attractive to prescribers. Tapentadol is a newer drug with similar dual action, although it is a more potent μ-opioid receptor agonist than tramadol.
Other compounds also have regulatory roles in the spinal cord. The cannabinoid receptors and the endogenous cannabinoids have recently become a focus for research on pain regulation. There are two cannabinoid receptors, both of which are G protein-coupled: CB1, expressed in the brain, spinal cord, and sensory neurons; and CB2, largely expressed in nonneural tissues, especially immune cells including microglia. Several endogenous cannabinoids have been identified, including members of the anandamide and 2-arachidonylglycerol (2AG) families. A combination of anecdotal evidence and clinical trial data suggests that cannabinoids may have an analgesic effect in patients with AIDS neuropathy or multiple sclerosis. Selective cannabinoid pathway agents under development, such as CB1 or CB2 agonists or inhibitors of endocannabinoid metabolism, may prove useful for pain management.

The pain processing circuit described above is responsible for producing acute nociceptive pain, a physiologic, adaptive sensation elicited only by noxious stimuli that acts as a warning or protective signal. There are some clinical situations, such as acute trauma, labor, or surgery, in which it is necessary to control nociceptive pain. In these cases, the pain pathway can be interrupted either by blocking transmission with local anesthetics (see Chapter 12) or by administering high-dose opioids. The opioids may be rapidly acting, such as remifentanil for intraoperative use, or more slowly acting, such as morphine; administered perioperatively, morphine retains activity for postoperative pain control.
Both peripheral inflammation and nervous system damage produce pain that is characterized by hypersensitivity to noxious and innocuous stimuli and by spontaneous pain that arises in the absence of any obvious stimulus. Understanding the mechanisms responsible for these types of clinical pain will facilitate both the appropriate use of currently available drugs and the development of novel therapeutic agents.
The ideal treatment of pain would be based on identifying and targeting the precise pain mechanisms operative in a particular patient and on normalizing abnormal pain sensitivity. Clinical pain syndromes may involve a combination of mechanisms, however, and there are few diagnostic tools to identify which specific mechanisms are responsible in a particular patient. Chronic pain conditions can be complicated to treat, and effective treatment usually demands multiple drugs (polypharmacy) to obtain the optimal therapeutic effect while reducing adverse effects. Chronic inflammatory pain conditions require the use of drugs that reduce the inflammatory response; such drugs may both correct the underlying inflammatory condition (disease-modifying therapy) and reduce the pain. For example, the nonsteroidal anti-inflammatory drugs (NSAIDs) (see Chapter 43, Pharmacology of Eicosanoids) are the first line of treatment for rheumatoid arthritis. By reducing inflammation, this intervention can decrease the release of chemical ligands that sensitize peripheral nerve terminals and thereby prevent peripheral sensitization (see below). Other disease-modifying anti-inflammatory treatments that may also reduce pain include cytokine inhibitors, sequestering agents such as TNF-α inhibitors, and immunosuppressants, as well as anti-nerve growth factor (NGF) monoclonal antibodies that are now under development.
The major agents used to treat most noninflammatory neuropathic or dysfunctional pain conditions are generally not disease-modifying because the underlying disease processes are either not known (e.g., fibromyalgia) or refractory to currently available treatments (e.g., neuropathic pain). Neuropathic pain associated with peripheral nervous tissue damage, spinal cord injury, or stroke commonly requires the use of several agents to alleviate pain symptoms. In nonmalignant pain, opioids have generally been used as a matter of last resort because of their adverse effects and because of the potential for the development of tolerance and physical dependence (see Chapter 19, Pharmacology of Drugs of Abuse). However, in recent years, opioids have increasingly been used for the management of chronic noncancer pain, albeit with the risks of producing drug-seeking behavior and creating opportunity for diversion of the drugs for illicit use. The U.S. Centers for Disease Control and Prevention (CDC) reported that opioid analgesics had a role in 71.3% of the 22,767 deaths due to prescription drug overdose occurring in the U.S. in 2013.
Severe acute pain due to trauma, surgery, or inflammation is usually treated with opioids and NSAIDs. The many available opioid agents permit a great deal of flexibility in both potency and duration of action. Remifentanil, a high-potency opioid, is cleared over the course of a few minutes, while methadone after prolonged use may be cleared over the course of days. Combinations of intermediate- and long-acting opioids may be used to tailor an analgesic regimen for pain that fluctuates in intensity over days, hours, or minutes. Remifentanil was administered during JD’s surgical debridement for optimal control of intraoperative pain, followed by a morphine bolus and infusion for postoperative pain control. The nausea and sedation produced in many patients by opioids are potential problems when these agents are used for day-surgery cases. Acute inflammatory pain conditions, such as pancreatitis, are often treated with morphine. Gout, a second example of an acute inflammatory condition producing severe pain, is usually treated with indomethacin (an NSAID) to reduce the pain rapidly, and more specific disease-modifying agents are used to correct the underlying disorder over the longer term (see Chapter 49, Integrative Inflammation Pharmacology: Gout).
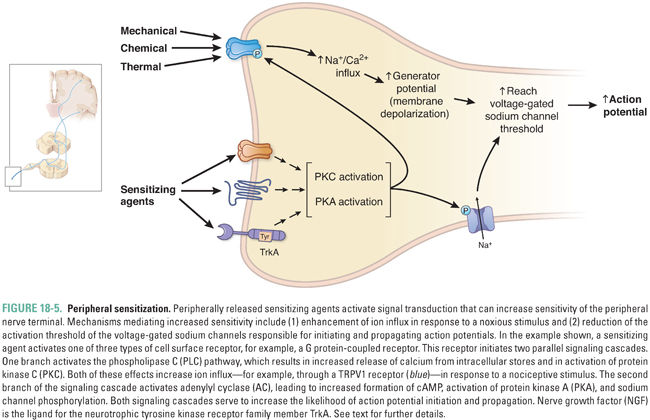
Several peripheral stimuli can cause primary afferent neurons to lower their activation thresholds and increase their responsiveness (Fig. 18-5). These alterations, which constitute peripheral sensitization, can result in allodynia, in which normally innocuous stimuli are perceived as painful, and hyperalgesia, in which high-intensity stimuli are perceived as more painful and longer lasting than usual at the site of injury (zone of primary hyperalgesia). Some inflammatory mediators released by injured (ATP) or immune (IL-1β) cells can directly activate nociceptors to signal to the CNS the presence of tissue injury and thereby evoke pain. The mechanisms responsible for primary hyperalgesia involve both direct changes in transduction and indirect changes induced by the release of effector molecules. An example of altered transduction is the change in heat activation of the TRPV1 receptor due to posttranslational modifications and altered membrane trafficking following activation of PKC and PI3K signaling pathways, which reduce the activation threshold of the receptor so that it can be activated by warm stimuli (38°–40°C) that are normally not painful. The major known effectors that produce peripheral sensitization are the inflammatory mediators bradykinin, the cytokine IL-1β, protons, histamine, prostaglandin E2, and NGF. Prostaglandin E2 acts on EP receptors, of which there are four types, while NGF acts on the neurotrophic tyrosine kinase receptor family member TrkA. The actions of histamine are more prominent on the subset of sensory neurons that contribute to itch.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


