INTRODUCTION
With over 10 billion neurons and an estimated 1014 synaptic connections, the human brain boasts unparalleled electrical complexity. Unlike myocardial tissue, where electrical signals spread synchronously through a syncytium of cells, proper functioning of the brain requires distinct isolation of electrical signals and thus demands a far higher level of regulation. Control of this complex function begins at the level of the ion channel and is further maintained through the effects of these ion channels on the activity of highly organized neuronal networks. Abnormal function of ion channels and neural networks can result in rapid, synchronous, and uncontrolled spread of electrical activity, which is the basis of a seizure.
A seizure can present with a variety of symptoms and result from a variety of causes. A single seizure should be distinguished from epilepsy, which refers to the condition in which an individual has a tendency toward recurrent seizures (i.e., a patient who has had a single seizure does not necessarily have epilepsy). Seizure symptoms vary according to the location of seizure activity and may include prominent motor symptoms and loss of consciousness (as seen in tonic–clonic seizures), paroxysmal alterations in nonmotor functions (e.g., sensation, olfaction, vision), or changes in higher order functions (e.g., emotion, memory, language, insight).
This chapter explores the molecular mechanisms by which the brain maintains precise control over the spread of electrical activity and how various abnormalities can undermine these physiologic mechanisms and lead to seizures. The various classes of antiepileptic drugs are then discussed, with an emphasis on molecular mechanisms for restoring inhibitory function in the brain and suppressing seizure activity.
 Jon arrives in the emergency department with his brother Rob at 9:12 PM. Because his brother is still too lethargic to speak, Jon relays most of the story to the attending physician. The two had been watching television when Jon noticed that his 40-year-old brother seemed to be daydreaming. Never missing an opportunity to tease, Jon began chiding his brother for “spacing out.” But instead of the boisterous laugh that he was so used to, Jon observed only a confused, almost fearful stare.
Jon arrives in the emergency department with his brother Rob at 9:12 PM. Because his brother is still too lethargic to speak, Jon relays most of the story to the attending physician. The two had been watching television when Jon noticed that his 40-year-old brother seemed to be daydreaming. Never missing an opportunity to tease, Jon began chiding his brother for “spacing out.” But instead of the boisterous laugh that he was so used to, Jon observed only a confused, almost fearful stare.
Jon recalls that his brother’s right hand suddenly began to bend into an awkward position and then to shake. The shaking grew worse, progressing gradually from the hand to the arm and then to the entire right side of the body. Jon then noticed that Rob’s body stiffened, almost as if he were attempting to contract every muscle in his body. This sustained contraction lasted for about 15 seconds and was followed by shaking movements of all four limbs that lasted another 30 seconds or so. The frequency of the shaking slowed after several minutes, and Rob then became limp, began breathing very heavily, and remained unresponsive. Rob regained consciousness on the way to the emergency department.
At the hospital, a magnetic resonance imaging (MRI) scan shows a small neoplasm in Rob’s left temporal lobe. Because the neoplasm appears to be benign, Rob, following the advice of his physician, decides not to undergo surgery. The potential benefits and risks of various antiepileptic drugs are discussed, including phenytoin, carbamazepine, valproic acid, and lamotrigine, and it is decided to start Rob on a regimen of carbamazepine to prevent further seizures.
Questions
1. By what mechanisms can a focal neoplasm result in a seizure?
2. Is there any clinical significance to the fearful, blank stare?
3. What is the significance of the order of spread of the seizure from the hand to the arm and then to the leg?
4. The generalized seizure that followed the right-sided shaking included a tonic phase (stiffening) followed by a clonic phase (shaking). What occurs at the molecular level to cause these symptoms?
5. Why was carbamazepine chosen as the antiepileptic treatment for Rob’s seizures?
 PHYSIOLOGY
PHYSIOLOGYThe normal human brain, in the absence of any lesions or genetic abnormalities, is capable of undergoing a seizure. Acute changes in the availability of excitatory neurotransmitters (e.g., caused by ingestion of the toxin domoate, which is a structural analogue of glutamate) or changes in the effect of inhibitory neurotransmitters (e.g., caused by injection of penicillin, a GABAA antagonist) can result in massive seizure activity in the otherwise healthy human brain. These examples demonstrate that the complex circuits within the brain exist in a balance between excitatory and inhibitory factors and that changes in either category of these control mechanisms can lead to major dysfunction.
In the CNS, two important elements normally involved in the fine-tuning of neuronal signaling also function to prevent the repetitious and synchronous firing characteristic of a seizure. At the cellular level, a “refractory period” induced by Na+ channel inactivation and K+ channel-mediated hyperpolarization prevents abnormal repetitive firing in neuronal cells. As discussed in Chapter 8, Principles of Cellular Excitability and Electrochemical Transmission, action potentials are propagated by voltage-sensitive ion channels. After initiation in the axon hillock, the action potential is propagated by alternating currents of depolarizing Na+ influx and hyperpolarizing K+ efflux. Throughout the course of an action potential (Fig. 16-1), the Na+ channels exist in three distinct states: (1) a closed state before activation, (2) an open state during depolarization, and (3) an inactivated state shortly after the peak of depolarization.
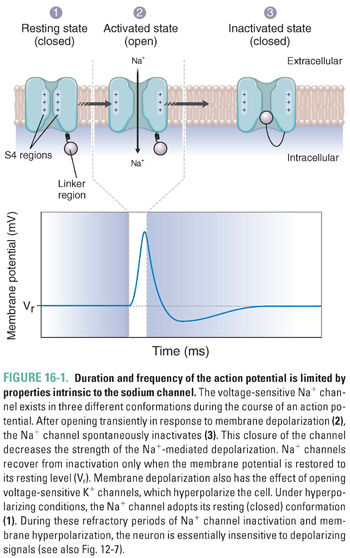
Because Na+ channels adopt the inactivated state in response to depolarization, action potentials are intrinsically self-limiting—Na+ channels will not recover from the inactivated state until the membrane is sufficiently repolarized. K+ channel opening repolarizes the cell, but the high K+ efflux transiently hyperpolarizes the membrane beyond its resting potential, further increasing the time before a new action potential can be generated. Thus, under physiologic conditions, the biochemical properties of Na+ and K+ channels impose a limit on the frequency of firing and help to prevent the repetitive firing characteristic of many seizure types.
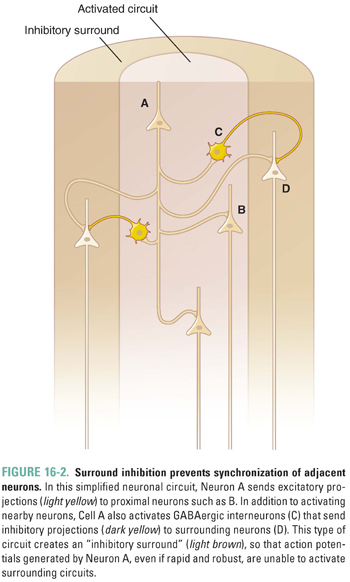
Beyond the single-cell level, neural networks ensure the specificity of neuronal signaling by restricting the effects of a given action potential to a defined area. Even a strong train of action potentials, if contained within about 1,000 neurons, will not generate seizure activity. This is quite a remarkable feat, given the close proximity of neurons in the CNS and the fact that a single neuron in the neocortex may have more than 1,000 postsynaptic connections. As seen in the simplified neural network in Figure 16-2, the firing neuron activates immediately neighboring neurons in addition to interneurons that transmit inhibitory (GABA) signals to surrounding neurons. This contrast of local amplification and surrounding cell inhibition results in what is referred to as surround inhibition. Surround inhibition is essential to the normal function of the nervous system, because this phenomenon not only amplifies local signals but also provides insulation and protection against synchronicity in surrounding areas. Many seizure disorders appear to result from disruption of this intricate balance.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGYBecause the pathophysiologic mechanisms underlying seizure disorders are only beginning to be determined, seizures are in part classified according to their clinical manifestations. There has been a tendency to consider seizures as a dichotomous process involving the whole brain or part of the brain and referred to as generalized or focal accordingly. This is probably an oversimplification. In fact, a seizure can involve a neuronal network that is restricted to one hemisphere or that gradually or rapidly involves both hemispheres, and seizures involving both hemispheres may be asymmetric and may not involve the whole cortex. Thus, the term generalized should not be taken to mean “the whole brain” but rather to indicate that the involved neuronal network is bilaterally distributed.
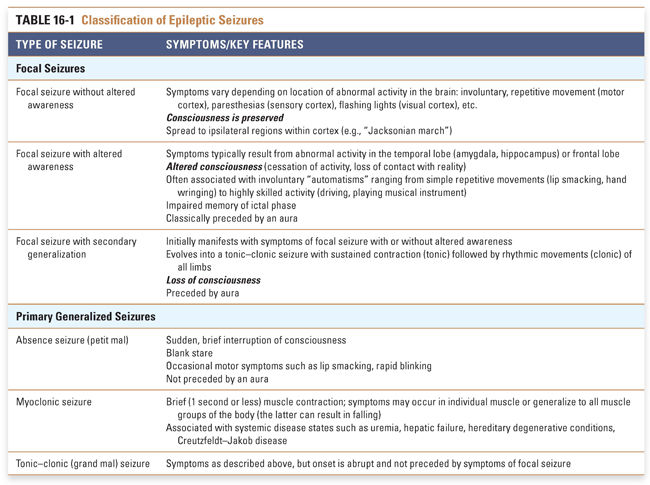
When seizures involve part of the brain, they are said to be focal. Depending on the location and size of the seizure focus, there may be alteration in awareness, in which case the seizure is referred to as focal dyscognitive or “focal with alteration in awareness.” (Note that this roughly corresponds to the previously termed complex partial seizure, a label that has been abandoned). In addition to helping the clinician define the underlying neuroanatomy, these associated symptoms have implications for the extent of disability and for defining appropriate treatments, including surgery (Table 16-1).
Whether a seizure involves one hemisphere or two, all seizures share the common characteristic of abnormal synchronous discharges. For this to occur, protective mechanisms must be compromised at the cellular and network levels. The direct causes of these changes can be primary (e.g., genetic abnormalities such as ion channel defects), secondary (e.g., changes in the neuronal environment induced by toxins, autoantibodies, or acquired lesions such as strokes or neoplasms), or a combination of the two (e.g., febrile seizures in children).
Pathophysiology of Focal Seizures
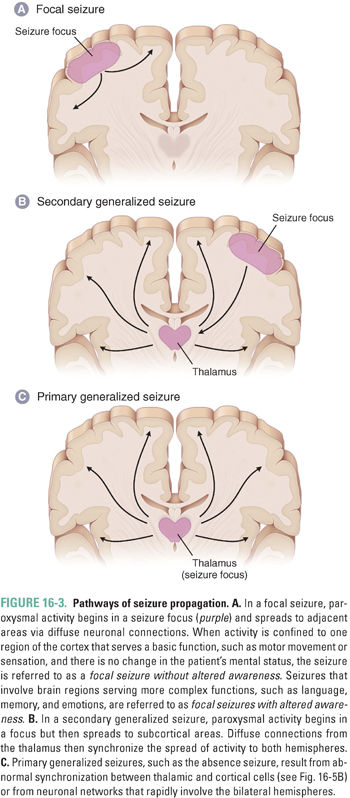
The focal seizure (Fig. 16-3A) occurs in three specific steps: (1) initiation at the cellular level by an increase in electrical activity, (2) synchronization of surrounding neurons, and (3) spread to adjacent regions of the brain. Seizures are initiated by a sudden depolarization within a group of neurons. This sudden change, called a paroxysmal depolarizing shift (PDS), lasts up to 200 ms and results in the generation of an abnormally rapid train of action potentials. Changes in the extracellular milieu, attributable, for instance, to a space-occupying lesion (such as in the introductory case), can have major effects on neuronal burst activity. For example, the space-occupying lesion could cause an increase in extracellular K+, which would blunt the effects of K+-mediated after-hyperpolarization by decreasing the magnitude of the K+ gradient between the outside and inside of the cell. Similarly, an increase in excitatory neurotransmitters or modulation of excitatory receptors by other exogenous molecules could increase burst activity. Increased burst activity could also result from properties intrinsic to the cell, such as abnormal channel conductance or altered membrane characteristics.
Because of surround inhibition, local discharges are often contained within a so-called focus and do not induce symptomatic pathology. These local discharges can be seen on an electroencephalogram (EEG) as sharp interictal spikes. Identification of these spikes can be useful in locating the seizure focus in a patient who is not actively undergoing a seizure. There are several pathways, however, whereby the seizure focus can override surround inhibition. Repetitive firing of neurons increases extracellular K+. As described above, this weakens K+-mediated hyperpolarization, allowing the seizure activity to spread. Rapidly firing neurons also open depolarization-sensitive NMDA channels (see Chapter 13, Pharmacology of GABAergic and Glutamatergic Neurotransmission) and accumulate Ca2+ in their synaptic terminals, both of which increase the likelihood of signal propagation and local synchronization. In many cases, it appears that the most important compromise of surround inhibition occurs at the level of GABAergic transmission. Decreases in GABA-mediated inhibition—because of exogenous factors, degeneration of GABAergic neurons, or changes at the receptor level—are major factors that aid in the synchronization of a seizure focus.
If the synchronizing focus is sufficiently strong, the abnormal, synchronized firing from a small neural network will begin to spread to neighboring regions of the cortex. During this spread to neighboring areas, the patient may experience an aura, a conscious “warning” of the spread of the seizure. In the introductory case, Rob’s aura manifested as a blank, fearful stare. Although the aura is usually stereotypical for a given patient, a wide variety of auras exist. These include a sense of fear and confusion, disturbances of memory (e.g., déjà vu) or language, altered sensations, or an olfactory hallucination. As the seizure continues to spread, it can lead to additional clinical manifestations; the specific manifestation depends on the brain regions that become involved. In the introductory case, the clinical symptoms initially started with shaking of the hand and progressed to the arm and then to the leg. This is a Jacksonian march (named after the English neurologist Hughlings Jackson, who first described the symptoms), where the clinical symptoms result from spread of synchronous activity across the motor homunculus.
Pathophysiology of Secondary Generalized Seizures
Focal seizures may become generalized by spreading along diffuse connections to involve both cerebral hemispheres. This is known as a secondary (or secondarily) generalized seizure (Fig. 16-3B). Typically, seizures spread to distant sites by following normal circuits, and this spread can occur through several pathways. U fibers connect various regions of the cortex; the corpus callosum allows for spread between hemispheres; and thalamocortical projections provide a pathway for diffuse synchronized spread throughout the brain. Once seizure activity spreads to involve both hemispheres, a patient usually loses consciousness.
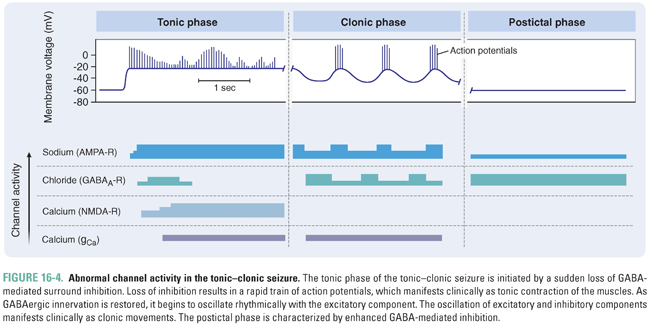
Among the secondarily generalized seizures, the tonic–clonic subtype is the most common. In the introductory clinical case, Rob underwent a period where he appeared to be contracting every muscle in his body, followed by an episode of uncontrolled shaking of all four limbs. These clinical symptoms can be understood at the level of abnormal channel activity (Fig. 16-4). The initial phase of the tonic–clonic seizure is associated with a sudden loss of GABA input, which leads to a long train of firing lasting for several seconds. This sustained, rapid firing manifests clinically as contraction of both agonist and antagonist muscles and is referred to as the tonic phase. Eventually, as GABA-mediated inhibition begins to be restored, AMPA-mediated and NMDA-mediated excitation starts to oscillate with the inhibitory component. This oscillatory pattern (when involving the motor cortex) results in clonic or shaking movements of the body. With time, the GABA-mediated inhibition prevails, and the patient becomes flaccid and remains unconscious during the postictal period until normal brain function returns.
Pathophysiology of Primary Generalized Seizures
Primary generalized seizures differ from focal seizures in both pathophysiology and etiology (Fig. 16-3C). In contrast to the focal seizure, where synchronicity begins with sudden trains of action potentials within an aggregate of neurons and subsequently spreads to adjacent regions, the primary generalized seizure emanates from central brain regions and then spreads rapidly to both hemispheres. These seizures do not necessarily begin with an aura (which is an important method of clinically distinguishing primary generalized seizures from focal seizures that secondarily generalize).
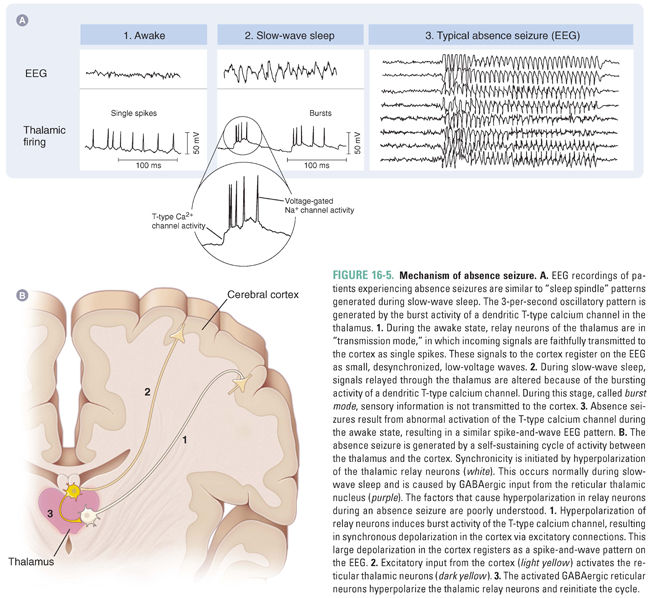
Currently, the best understood of the primary generalized seizures is the absence seizure (also known as the petit mal seizure). Absence seizures are characterized by sudden interruptions in consciousness that are often accompanied by a blank stare and occasional motor symptoms, such as rapid blinking and lip smacking. Absence seizures are thought to result from abnormal synchronization of thalamocortical and cortical cells. The underlying pathophysiology of absence seizures is based on the observation that patients experiencing absence seizures have EEG readings somewhat similar to the patterns generated during slow-wave (stage 3) sleep.
Relay neurons connecting the thalamus to the cortex exist in two different states depending on the level of wakefulness (Fig. 16-5A). During the awake state, these neurons function in transmission mode, whereby incoming sensory signals are faithfully transmitted to the cortex. During sleep, however, the transient, bursting activity of a unique, dendritic T-type calcium channel alters incoming signals so that output signals to the cortex have an oscillatory firing rate, which, on an EEG, has a characteristic “spike and wave” readout. In this slow-wave sleep state, sensory information is not transmitted to the cortex.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


