Modern pharmacologic agents are used to treat or control diseases that range from hypertension to human immunodeficiency virus (HIV) infection. In many cases, the same drug regimens are used for populations of people. Large variations among individuals are often found in response to drug therapy, however. These variations range from potentially life-threatening adverse drug reactions to equally serious lack of therapeutic efficacy. Many factors influence the drug response phenotype, including age, gender, and underlying disease, and genetic variation plays an important role. Interindividual differences in the genes that encode drug targets, drug transporters, and enzymes that catalyze drug metabolism can profoundly affect the success or failure of pharmacotherapy.
Pharmacogenetics is the study of the role of inheritance in variation in drug response. The convergence of recent advances in genomic science and equally striking advances in molecular pharmacology has resulted in the evolution of pharmacogenetics into pharmacogenomics. The promise of pharmacogenetics-pharmacogenomics is the possibility that knowledge of a patient’s DNA sequence could be used to enhance pharmacotherapy, maximize drug efficacy, and reduce the incidence of adverse drug reactions. Therefore, pharmacogenetics and pharmacogenomics represent an important aspect of the aspiration to “personalize” or “individualize” medicine—in this case, drug therapy. This chapter describes the principles of pharmacogenetics and pharmacogenomics as well as recent developments in this discipline. Several key examples are cited in which knowledge of pharmacogenetics-pharmacogenomics may help to individualize drug therapy.
 Robert H, a 66-year-old man, is shoveling snow one wintry morning in Minnesota when he slips and falls on a patch of ice. He immediately feels pain in his left hip and is unable to stand. He is brought to the hospital, where x-rays reveal that he has fractured his hip. He undergoes surgery the next day and is discharged to a rehabilitation hospital 3 days later. After less than 24 hours at the rehabilitation hospital, Mr. H develops the sudden onset of pleuritic chest pain. He is brought to the emergency department, where a computed tomography (CT) scan with intravenous contrast reveals a pulmonary embolus. He is treated with heparin and is anticoagulated with warfarin at a starting dose of 5 mg each day, with a target international normalized ratio (INR) of 2.0–3.0. Mr. H is discharged back to his rehabilitation hospital and referred to his local physician. When the INR is subsequently measured, it is 6.2, a value associated with an increased risk of hemorrhage. He is taking no other medication that might interfere with plasma levels of warfarin. The physician advises Mr. H to stop taking warfarin for 2 days. After multiple attempts at adjusting his dose of warfarin, Mr. H eventually reaches a stable INR of 2.5 when taking 1 mg of warfarin each day.
Robert H, a 66-year-old man, is shoveling snow one wintry morning in Minnesota when he slips and falls on a patch of ice. He immediately feels pain in his left hip and is unable to stand. He is brought to the hospital, where x-rays reveal that he has fractured his hip. He undergoes surgery the next day and is discharged to a rehabilitation hospital 3 days later. After less than 24 hours at the rehabilitation hospital, Mr. H develops the sudden onset of pleuritic chest pain. He is brought to the emergency department, where a computed tomography (CT) scan with intravenous contrast reveals a pulmonary embolus. He is treated with heparin and is anticoagulated with warfarin at a starting dose of 5 mg each day, with a target international normalized ratio (INR) of 2.0–3.0. Mr. H is discharged back to his rehabilitation hospital and referred to his local physician. When the INR is subsequently measured, it is 6.2, a value associated with an increased risk of hemorrhage. He is taking no other medication that might interfere with plasma levels of warfarin. The physician advises Mr. H to stop taking warfarin for 2 days. After multiple attempts at adjusting his dose of warfarin, Mr. H eventually reaches a stable INR of 2.5 when taking 1 mg of warfarin each day.
Questions
1. What molecular mechanisms could be responsible for the apparent sensitivity of Mr. H to warfarin?
2. What additional laboratory information could assist in anticoagulating this patient?
3. Would that information have helped in the selection of Mr. H’s initial warfarin dose?

Genomic Variation and Pharmacogenomics
The human genome contains approximately 3 billion nucleotides. According to current estimates, the genome contains approximately 19,000–20,000 protein-coding genes that, through alternative splicing and post-translational modification, may encode 100,000 or more proteins. On average, any two people differ at about one nucleotide in every 1,000 in their genome, totaling an average interindividual difference of 3 million base pairs throughout the genome. The majority of these differences are so-called single nucleotide polymorphisms or SNPs (pronounced “snips”), in which one nucleotide is exchanged for another at a given position. SNPs and other differences in DNA sequence can occur anywhere in the genome, in both coding regions and noncoding regions. If a SNP changes the encoded amino acid, it is called a nonsynonymous coding SNP (cSNP). The remaining differences in DNA sequence involve insertions, deletions, duplications, and reshufflings, sometimes of just one or a few nucleotides but occasionally of whole genes or larger DNA segments that include many genes. Functionally significant DNA sequence differences that we currently understand tend to fall within genes, either within their coding sequences or in the promoters, enhancers, splice sites, or other sequences that control gene transcription or mRNA stability. Heritable genetic regulation that does not occur through DNA sequence changes, termed epigenetics, also contributes to functionally significant variation in genes and gene expression. Taken together, these differences constitute each person’s genetic individuality. Some of that individuality affects the way in which each person will respond to drug treatment.

The concept that inheritance might be an important determinant of individual variation in drug response emerged half a century ago. It originally grew out of clinical observations of striking differences among patients in their response to “standard” doses of a drug. Those observations, coupled with twin and family studies that showed inherited variations in plasma drug concentrations and other pharmacokinetic parameters, led to the birth of pharmacogenetics. Many of those original examples of pharmacogenetic variation, and many of the most striking examples even today, involve pharmacokinetics—factors that influence the concentration of drug reaching its target(s). However, examples of pharmacogenetic variation in the drug target, so-called pharmacodynamic factors, are also being reported with increasing frequency. Pharmacogenetic variation related to unpredicted (idiosyncratic) adverse effects from drugs has also been described. While not detailed further in this chapter, an example of an idiosyncratic pharmacogenetic effect is provided by the serious hypersensitivity reaction associated with the HLA-B*5701 allele in patients taking abacavir, a reverse transcriptase inhibitor and anti-HIV agent (see Chapter 38, Pharmacology of Viral Infections). Predrug testing for HLA-B*5701 has been shown to nearly eliminate such hypersensitivity reactions.
Variation in Enzymes of Drug Metabolism: Pharmacokinetics
Inherited variation in enzymes that catalyze drug metabolism is the most common factor responsible for pharmacogenetic variation in response to medications. The enzymes involved in drug metabolism are discussed in Chapter 4, Drug Metabolism. There are two broad categories of drug-metabolizing enzymes: those that catalyze phase I reactions (functionalization reactions that typically involve oxidation or reduction) and those that catalyze phase II reactions (typically, conjugation reactions that add groups, such as glucuronic acid, that enhance drug solubility and thus drug excretion). Phase I and phase II reactions do not necessarily occur in that order, and metabolic intermediates resulting from both types of reactions may be pharmacologically active. In fact, some medications are administered as inactive prodrugs that must undergo phase I and/or phase II metabolism before they can exert their pharmacologic effect.
Genetic polymorphisms are common in enzymes that catalyze drug metabolism, and clinically significant polymorphisms have been found in nearly all of the major enzymes involved in both phase I and phase II reactions (Table 7-1). Two “classic” examples are provided by the inherited variations in the enzymatic hydrolysis of the short-acting muscle relaxant succinylcholine by the enzyme butyrylcholinesterase (BChE; also known as serum cholinesterase) and the enzymatic acetylation of drugs such as the antituberculosis drug isoniazid (see Chapter 35, Pharmacology of Bacterial and Mycobacterial Infections: Cell Wall Synthesis). Patients with variations in BChE have a decreased rate of metabolism of succinylcholine, resulting in prolonged paralysis after drug exposure. A genetically polymorphic phase II enzyme, N-acetyltransferase 2 (NAT2), catalyzes the acetylation of isoniazid. Patients treated with isoniazid can be classified as either “slow acetylators,” who metabolize isoniazid slowly and have high blood drug levels, or “fast acetylators,” who metabolize isoniazid rapidly and have low blood drug levels. Family studies have shown that the rate of isoniazid biotransformation is inherited.
The slow-acetylator phenotype is associated with drug toxicities that result from excessive drug accumulation; examples include hydralazine- and procainamide-induced lupus and isoniazid-induced neurotoxicity and liver injury. Although the antihypertensive agent hydralazine is rarely used today in the treatment of hypertension, this drug has recently reemerged as one of the two active components in BiDil, a combination drug approved for the treatment of patients with symptomatic heart failure. It is of interest that the US Food and Drug Administration (FDA) has approved BiDil for use only in patients of African ancestry, presumably because of an ethnically dependent genetic difference in response to this drug.
Early examples of the importance of pharmacogenetic variation, such as those represented by BChE and NAT2, served as a stimulus to search for additional examples. Most of the second-generation examples continued to be associated with pharmacokinetics and continued to be recognized from clinical observations—often from adverse drug responses. They were most often studied either by administering a “probe drug” to a group of subjects and measuring plasma or urinary drug and/or metabolite concentrations or by directly assaying a drug-metabolizing enzyme in an easily accessible tissue such as the red blood cell (e.g., a series of methyltransferase enzymes). Two prototypic examples that have become pharmacogenetic “icons” are the cytochrome P450 2D6 (CYP2D6) and thiopurine S-methyltransferase (TPMT) genetic polymorphisms. Because of the clinical implications of these polymorphisms, the FDA in its 2003 “Guidance on Pharmacogenomic Data” cited CYP2D6 and TPMT as examples of valid pharmacogenomic biomarkers.
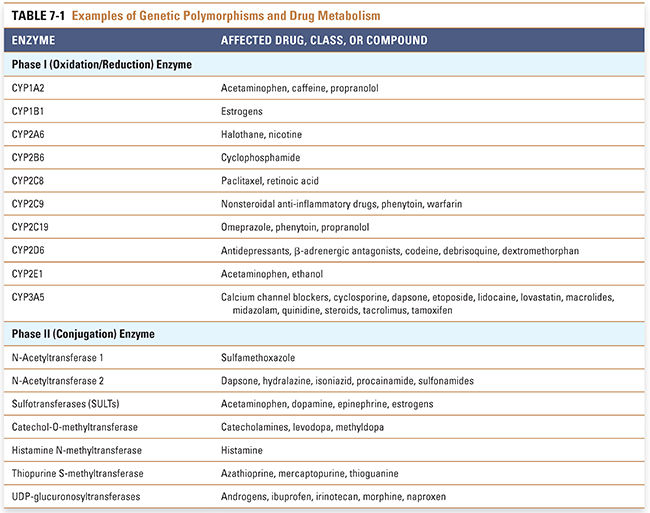
CYP2D6 is a member of the cytochrome P450 (CYP) family of microsomal, phase I drug-metabolizing enzymes. CYP2D6 contributes to the metabolism of a large number of medications, including antidepressants, antipsychotics, antiarrhythmics, and analgesics. The CYP2D6 polymorphism was originally described by two different laboratories studying two different probe drugs: the antihypertensive debrisoquine and the oxytocic agent sparteine. The frequency distribution of the debrisoquine urinary metabolic ratio, the ratio of the parent drug to its oxidized metabolite, is shown in Figure 7-1A for a Northern European population. Shown at the far right of the figure is a group of “poor metabolizers” of debrisoquine, subjects homozygous for recessive alleles (genes) coding for enzymes with decreased activity or for deletion of the CYP2D6 gene; shown in the middle is the large group of “extensive metabolizers,” subjects heterozygous or homozygous for the “wild-type” allele; and shown at the far left is a small subset of “ultrarapid metabolizers,” some of whom have multiple copies of the CYP2D6 gene.
Several molecular genetic mechanisms are responsible for variation in CYP2D6 enzyme activity, including nonsynonymous cSNPs, gene deletion, and gene duplication; some ultrarapid metabolizers can have up to 13 copies of the gene. It has been estimated that 6% to 10% of Caucasians are CYP2D6 poor metabolizers. Among East Asians, in contrast, the poor-metabolizer phenotype is present at a frequency of just 1% to 2%. The ultrarapid-metabolizer phenotype, rare in most Caucasian populations, has a frequency of 3% in Spaniards and up to 13% in Ethiopians. These ethnic differences have potentially important medical implications because CYP2D6 metabolizes many commonly prescribed medications, including the β-adrenergic blocker metoprolol, the neuroleptic haloperidol, the opioids codeine and dextromethorphan, and the antidepressants fluoxetine, imipramine, and desipramine, among many others (Table 7-1). Therefore, poor metabolizers for CYP2D6 can potentially experience an adverse drug effect when treated with standard doses of agents such as metoprolol that are inactivated by CYP2D6, whereas codeine is relatively ineffective in poor metabolizers because it requires CYP2D6-catalyzed metabolism to form the more potent opioid morphine. Conversely, ultrarapid metabolizers may require unusually high doses of drugs that are inactivated by CYP2D6, but those same subjects can be “overdosed” with codeine, suffering respiratory depression or even respiratory arrest in response to “standard” doses. In one tragic case, a nursing infant whose mother was an ultrarapid CYP2D6 metabolizer died when the mother was prescribed a standard dose of codeine and the baby was overdosed on morphine present in the breast milk. For ultrarapid and poor metabolizers, alternative analgesics are now recommended as substitutes for codeine.
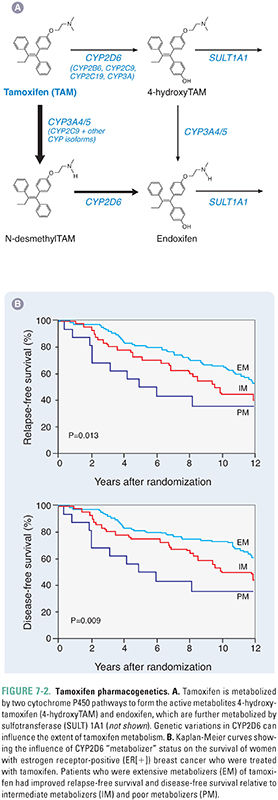
CYP2D6 genetic polymorphisms are also important for the efficacy of the breast cancer drug tamoxifen. Tamoxifen is used to block the estrogen receptor (ER) in approximately 60% of breast cancer patients with ER-positive tumors. However, tamoxifen is a prodrug that requires metabolic activation to form 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen (endoxifen) (Fig. 7-2A). These metabolites are approximately 100 times more potent as antagonists of the ER than the parent drug. As a result, patients who are CYP2D6 poor metabolizers (Fig. 7-1) are relatively unable to form the active 4-hydroxy metabolites of tamoxifen. Poor-metabolizer patients have worse outcomes with respect to breast cancer recurrence than do CYP2D6 extensive metabolizers (EMs) (Fig. 7-2B). Furthermore, if EM patients are co-administered other drugs such as antidepressants that are good CYP2D6 substrates, they may receive less benefit from tamoxifen therapy than do CYP2D6 EMs who are not co-administered drugs that compete with tamoxifen for CYP2D6-catalyzed metabolism.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


