Figure 4.1
Common pedigree symbols, definitions, and abbreviations. From Bennett R, French K, Resta R, Doyle D. Standardized human pedigree nomenclature: update and assessment of the recommendations of the national society of genetic counselors. J Genet Couns. 17;2008:424–33 [2]. Reprinted with permission from Springer.
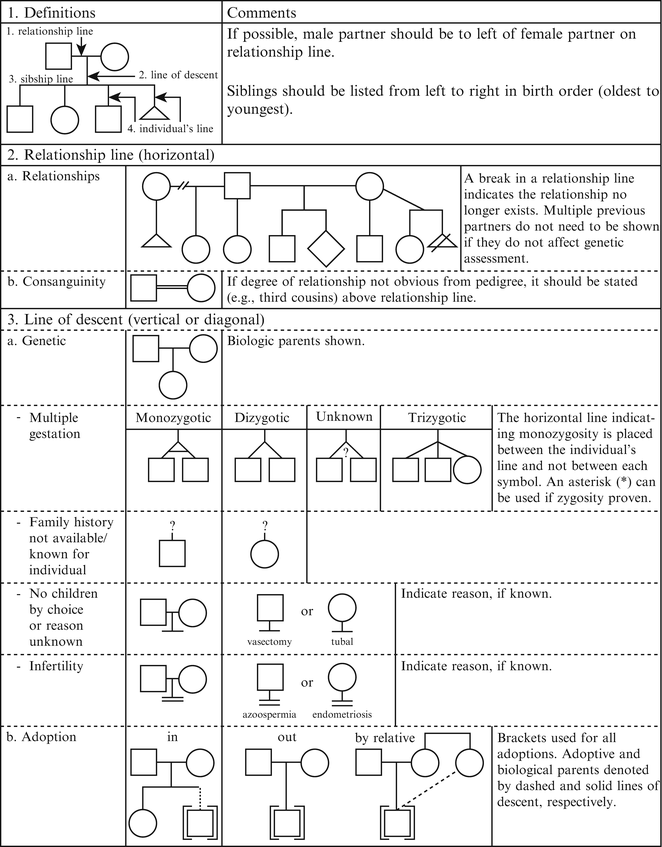
Figure 4.2
Pedigree line definitions. From Bennett R, French K, Resta R, Doyle D. Standardized human pedigree nomenclature: update and assessment of the recommendations of the national society of genetic counselors. J Genet Couns. 17;2008:424–33 [2]. Reprinted with permission from Springer
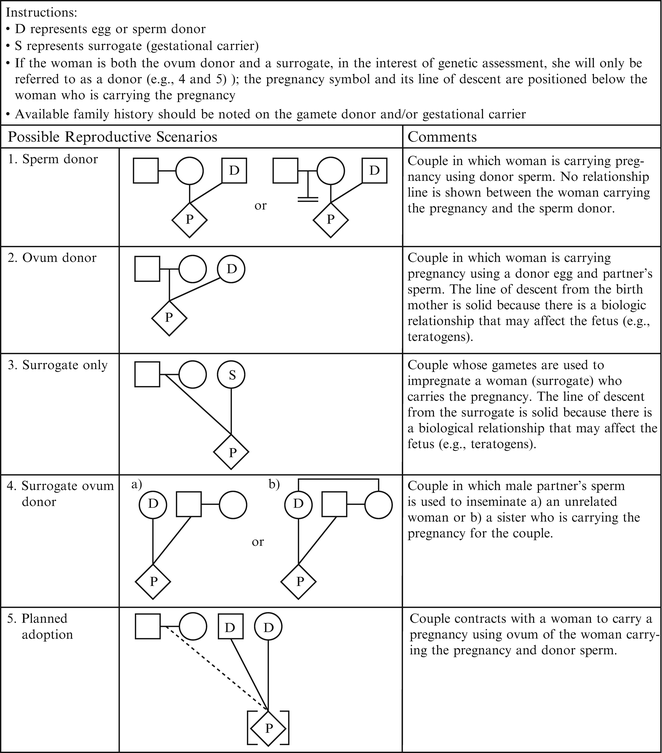
Figure 4.3
Assisted reproductive technology symbols and definitions. From Bennett R, French K, Resta R, Doyle D. Standardized human pedigree nomenclature: update and assessment of the recommendations of the national society of genetic counselors. J Genet Couns. 17;2008:424–33 [2]. Reprinted with permission from Springer
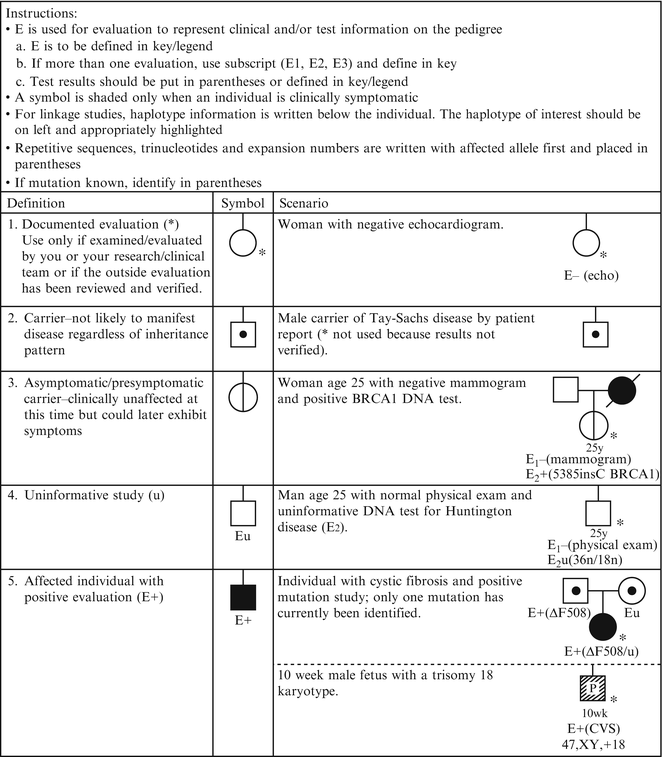
Figure 4.4
Pedigree symbols of genetic evaluation/testing information. From Bennett R, French K, Resta R, Doyle D. Standardized human pedigree nomenclature: update and assessment of the recommendations of the national society of genetic counselors. J Genet Couns. 17;2008:424–33 [2]. Reprinted with permission from Springer
Pedigrees now form the cornerstone for determination of diagnosis, pattern of inheritance, and recurrence risk. Visually recording elements of family and medical history in the form of a pedigree serves many purposes including: user orientation (to family relationships, source of the information included and reason for pedigree construction), improved readability, risk assessment, validation of information included, compliance with medical documentation standards, communication, and patient education. Well-constructed pedigrees also may result in cost savings by describing evaluations already performed to avoid duplicate testing, and documenting familial mutations necessary for the most cost-effective testing of family members. In addition, collection of family medical information has aided in the understanding of many unique features of hereditary disorders, including natural history, variability, and gene-gene or gene-environment interactions.
Collection of a family pedigree represents an opportunity to build a relationship with the patient and family and to learn about how the family functions. As the genetic counselor or other healthcare provider explains the purpose of obtaining family history, an atmosphere of open communication and respect can be established. This process provides a window to the social relationships and psychosocial and educational needs of patients and families. In the social sciences, genograms are used to graphically depict family dynamics that influence individual behaviors [3]. This information also is essential for successful counseling of patients in the clinical genetics setting, and while not always recorded in the same fashion, family dynamics are a vital part of the process of pedigree gathering. Observations about coping mechanisms, assumptions about disease causation, family hierarchy, key life experiences, stress levels, body language, and religious and ethnic influences all are integrated into consideration about the most effective ways to communicate information about a diagnosis, prognosis, or management plan to patients and families.
Ideally, the pedigree is collected in a face-to-face session. This is usually done prior to or at the beginning of the clinical genetics evaluation, but may be done later, particularly when evaluating a pregnancy or a newborn with an unanticipated, newly diagnosed condition. Advance notice to patients and their families about the nature of information to be collected can facilitate the accuracy and completeness of the information provided by the family. In addition, electronic tools such as the US Surgeon General’s Family Health Portrait tool (http://www.hhs.gov/familyhistory) are available to engage patients in collecting their family health information.
At a minimum, a three-generation pedigree should be collected, including all first-degree relatives (parents, children, full siblings), second-degree relatives (grandparents, aunts, uncles, nieces and nephews, half-siblings, grandchildren), and, as pertinent, many third-degree relatives (cousins, great-aunts, great-uncles, great-grandparents). This group can be expanded or condensed, depending on the nature of the referral and patient responses to preliminary questioning about features relevant to the reason for referral. For example, genetic evaluations for hereditary cancer syndromes may necessitate a more extended family pedigree, while a brief, focused pedigree may suffice when discussing cystic fibrosis carrier testing.
Information that should be collected about each individual in the pedigree is listed in Table 4.1. Modifications may be needed depending on the nature of the diagnosis under investigation; recommendations from the NSGC detail additional questions appropriate for individuals being evaluated for hereditary cancer syndromes [4]. Because family medical histories change over time, pedigrees should be updated as new information is learned. Ethnicity, consanguinity, and unique biological relationships should be recorded using standard notation. All reported diagnoses or conditions ideally should be confirmed through authorized request and review of medical records. Key records to obtain include pathology reports, test results (particularly for any genetic testing that has been performed), imaging reports, and autopsy reports. In the absence of these documents, family genealogies or death certificates may provide some degree of verification of reported information.
Table 4.1
Collection of family medical history: what to ask?
For all family members |
Current age; year of birth |
Exact relationship to the proband |
General physical and mental health status |
History of major acute or chronic illness, hospitalizations, and surgeries |
History of learning problems, diagnosed disabilities, or intellectual disability |
Highest grade level completed (when relevant) |
Employment (when relevant) |
Reproductive history, including pregnancies, miscarriages, elective terminations, infertility, and choice not to have children |
Gestational age and last menstrual period for ongoing pregnancies |
Consanguinity |
Ethnicity |
Targeted questions relevant to the reason for evaluation, for example, key symptoms or features of the condition in question, pertinent evaluations |
Age at death; year of death; cause of death |
For family member known to be affected by the condition in question |
Diagnosis |
Age at diagnosis or disease onset |
Method of diagnosis |
Evaluations and testing completed |
Symptoms |
Information about ongoing treatment or management plan |
Availability of medical records for review |
An important issue in the use of pedigrees for clinical evaluations and research is the issue of individual confidentiality [5]. Each member of the family has a right to expect that medical information will remain confidential. This becomes complicated when one considers the pedigree that may contain both reported (“hearsay”) and confirmed information for numerous individuals. Those people may have willingly shared information with the patient but may not want it shared with other family members. If subsequent to an evaluation a patient requests release of his or her pedigree to another family member, a provider should carefully consider the question of ownership of the pedigree information and be attuned to the potential consequences of releasing the (identifiable) information about other family members. Current interpretation of regulations outlined by HIPAA and other medical records privacy legislation may influence how such information is shared. Professional organizations including the American Society of Human Genetics also have developed position statements on this issue [6].
Patterns of Inheritance
One key use of the carefully collected and verified pedigree is determination of the most likely mode of inheritance of a condition in a family. This will have relevance to assessing recurrence risks, approaches to testing, and in some cases even prognosis. The concept of patterns of inheritance extends from the seventeenth-century work of Gregor Mendel, who described transmission of traits associated with single genetic loci. Transmission of human genetic conditions and traits has proven to be more complex, involving not only the single-gene patterns first described by Mendel but also chromosomal inheritance, mitochondrial inheritance, and multifactorial inheritance. Other genetic factors which can influence transmission of disease include imprinting, uniparental disomy, unstable DNA, gene-environment interactions, mosaicism, and synergistic heterozygosity. Undoubtedly, additional factors influencing transmission and expression of inherited traits will be elucidated as our understanding of the human genome expands. As of January 2012, the Online Mendelian Inheritance in Man (OMIM), a continuously updated catalog of human genes and genetic phenotypes, listed 13,775 identified genes and 4,520 genetic disorders for which the molecular basis is known [7]. Identified genetic disorders with known patterns of inheritance are commonly inherited as autosomal, X-linked, or mitochondrial.
Mendelian Inheritance Patterns
For a summary of Mendelian inheritance patterns, see Table 4.2.
Table 4.2
Features of Mendelian patterns of inheritance
Autosomal dominant |
Male-to-male transmission occurs; both sexes can transmit to offspring |
Affected family members in multiple generations; “vertical transmission” typically showing affected descendants of affected individuals and unaffected descendants of unaffected individuals |
Males and females affected, typically to comparable extent |
Variability of clinical findings |
Later/adult onset in some disorders |
Homozygotes may be more severely affected than heterozygotes |
Homozygosity may be lethal |
Occurrence of new mutations |
Nonpenetrance; apparent “skipping” of generations |
Germline mosaicism reported |
Autosomal recessive |
Affected family members are usually in one generation; “horizontal” inheritance |
Males and females equally likely to be affected; parental consanguinity or a small mating pool may influence disease occurrence |
Disease severity is usually consistent among affected family members |
Early onset of symptoms more typical |
New mutations rare |
May see higher frequency of disease in certain ethnic groups |
X-linked dominant |
No male-to-male transmission |
Affected females usually have milder symptoms than affected males |
Affected males have no affected sons, but all daughters will be affected |
May mimic autosomal dominant inheritance |
May be lethal in affected males; reflected by a paucity of males or overrepresentation of females in the pedigree |
Increased occurrence of miscarriage may be observed |
X-linked recessive |
No male-to-male transmission |
Males more frequently affected |
Carrier females usually unaffected but may have mild symptoms |
Affected males in a family are related through females |
Y-linked |
Male-to-male transmission only |
Association with increased rates of male infertility |
Discrepancy between chromosomal and phenotypic sex |
Autosomal Dominant Inheritance
In classic autosomal dominant inheritance, an affected individual has one non-functional or mutant allele at a particular locus, the presence of which causes disease. Each affected individual in a pedigree has a 50 % chance of passing the disease-associated mutation to each of his or her offspring. Additional genetic and non-genetic factors may influence the occurrence of these conditions in families. A key feature of autosomal dominant inheritance is the observance of male-to-male transmission of the condition or trait. Transmission from fathers to sons is not seen in X-linked dominant inheritance, which can be confused with autosomal dominant inheritance on first analysis. Table 4.2 lists additional features of autosomal dominant inheritance, and an example pedigree is shown in Fig. 4.5.
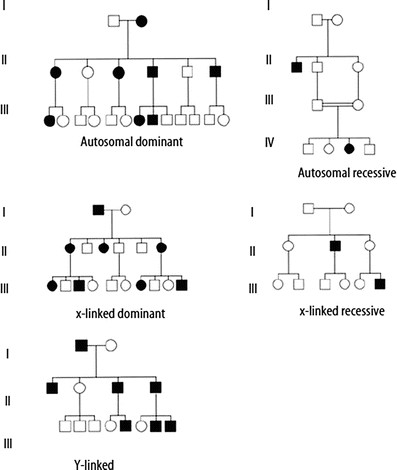
Figure 4.5
Example pedigrees for Mendelian patterns of inheritance
Autosomal Recessive Inheritance
In autosomal recessive inheritance, an affected individual has two non-functional or mutant alleles at a particular locus. Carriers of autosomal recessive conditions have one nonfunctional allele at the gene locus, but usually have no symptoms as they also have one normal, functional copy of the gene. If both partners of a couple are carriers of the same autosomal recessive condition, with each pregnancy there is a one in four (25 %) chance of having an affected child, a two-in-four (50 %) chance that a child will be a carrier and a one in four (25 %) chance a child will be neither a carrier nor affected. After birth, if a child of a carrier couple is not affected by the condition in question, he or she has a two-in-three chance of being a carrier. Risk to future offspring of a known carrier depends on the likelihood that his or her partner is also a carrier. This is influenced by the frequency of the disease, which may vary among different ethnic groups or populations. Features of autosomal recessive inheritance are listed in Table 4.2, and an example pedigree is shown in Fig. 4.5.
X-Linked Dominant Inheritance
In X-linked dominant inheritance, an affected individual has one nonfunctional or mutant allele at a locus on an X-chromosome. X-linked dominant conditions can occur in either males or females. Risk for offspring of an affected female is 50 %, regardless of the sex of the offspring. Risk to offspring of affected males is sex dependent, with all daughters but no sons inheriting the gene mutation. Many of these conditions, however, are lethal in males, so pedigrees may show overrepresentation of females or increased frequency of miscarriages, presumably of affected male fetuses (see Table 4.2 and Fig. 4.6).
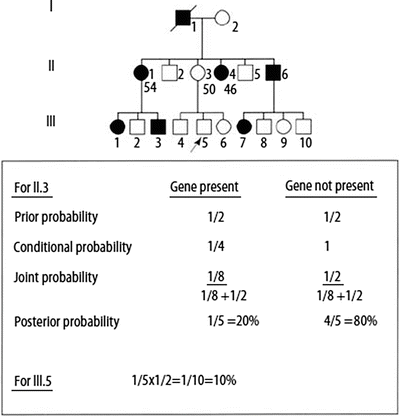
Figure 4.6
Bayesian analysis for risk assessment in an autosomal dominant, adult-onset hereditary cancer disorder. Ages of selected individuals in generation II are shown below the pedigree symbols
X-Linked Recessive Inheritance
Traditional X-linked recessive inheritance is characterized by occurrence of the condition in males having a non-functional or mutant allele for a gene on the X-chromosome. Affected males in the family will be related to each other through females. (See the pedigree in Fig. 4.5, and Table 4.2 for additional features.) Typically, carrier females are unaffected; however, due to lyonization (random inactivation of one X chromosome in each somatic cell in a female), carrier females may have mild symptoms. This occurs when, by chance, the X chromosome with the non-functional allele remains active in a majority of the cells within the critical tissue(s) for the disorder. The likelihood of symptoms in carrier females varies considerably among disorders. Risk to offspring of carrier females is 25 % overall, or 50 % for affected status if the fetus/offspring is male. Fifty percent of the female offspring of carrier females will also be carriers. Offspring of affected males will not be classically affected, but all daughters will be carriers.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


