Pathogenesis of Viral Infection
Mark T. Heise
Herbert W. Virgin
Introduction to Viral Pathogenesis
Viral pathogenesis is the series of steps that occurs when a virus infects the host. Viruses are obligate parasites of living cells that cannot live independent of an intricate relationship with an infected cell. The cells targeted by viruses during infection survive, differentiate, and function in a tissue that has an intimate relationship with other tissues and physiologic processes in the body. Thus, in the same sense that viruses are obligate parasites of living cells, they are obligate parasites of tissues in living organisms. Living in tissues and spreading from host to host represent processes only approximated in cell culture or biochemical experimentation. The study of viral pathogenesis elucidates this special relationship between the virus and the intact host.
The term pathogenesis refers to the processes related to disease induction; therefore, viral pathogenesis often refers to disease induction by a virus rather than the process of infection per se. However, viral infection does not always result in apparent or immediate disease, and the border between infection and disease becomes less clear as we learn more. It is most useful to consider the pathogenesis of infection independently of whether or not severe or immediate disease is induced. As the pathogenesis of infection is analyzed, the pathogenesis of disease can be considered as a subset of events that occur in vivo during infection. Fundamentally important mechanisms are revealed when one considers how the pathogenesis of infection differs between the host with asymptomatic infection or minimal disease and the host doomed to suffer severe consequences of viral infection.
Viral pathogenesis is the integrated result of many complex factors unique to a particular virus, a particular species, and an individual host. The interplay of these factors determines the nature of infection, whether disease occurs, and the severity of disease. In many cases, individual factors involved in pathogenesis can be studied in detail. For example, the crystal structure of a viral immune evasion protein allows predictions as to protein function during infection in vivo. However, definitive pathogenesis experiments often falsify such predictions. Thus, all conclusions derived from reductionist experiments utilizing molecular and biochemical approaches need to be validated in living organisms to understand viral pathogenesis.
The complexity of directly testing molecular mechanisms in vivo has been a significant stumbling block for pathogenesis research. The difficulty of doing definitive pathogenesis experiments that prove a molecular mechanism in vivo has resulted in pathogenesis research being relegated at times to a lesser status as a phenomenological science when compared to studies of molecular and biochemical mechanisms. However, in the end, infection occurs in the intact host; thus, events in vivo are highly relevant. With the development of new tools, in particular those that allow genetic analysis of pathogenesis from both the viral and host standpoint, pathogenesis research has rapidly evolved into a mechanistic science. Moreover, classical studies of pathogenesis have often stood the test of time and are useful at a minimum for defining the fundamental questions to be addressed using molecular pathogenesis approaches.
Goals of This Chapter
This chapter will focus on integrating classical concepts of pathogenesis with more current molecular understanding of viruses. Viral pathogenesis is the subject of many authoritative texts, and the reader is referred to one such text for detailed treatment of many of the important concepts developed here in necessarily shorter form.195,196 There is a rich history of how the initial observations that form the basis for our current understanding of viral pathogenesis were made.196 These founding studies are important to our understanding of the pathogenesis of infection as well as to major advances in human health such as the elimination of smallpox and the development of vaccines against diseases such as polio, measles, mumps, rubella, hepatitis B virus (HBV), and human papillomavirus. Exposure to the historical roots of pathogenesis research is strongly recommended for those who wish to delve into viral pathogenesis as a career or avocation.
Because it is impossible to provide a detailed description of the pathogenesis of all viral infections in a single chapter, the goal of this chapter is to provide a conceptual framework for understanding viral pathogenesis. Principles will be defined and then examples provided from a broad range of infections. For details of specific viral infections, refer to chapters on individual viruses. This chapter will specifically focus on the pathogenesis of infection with nucleic acid–containing viruses in mammals. There is a rich body of knowledge on plant viral diseases and prion diseases that will not be addressed in this chapter.
Definitions and Concepts in Viral Pathogenesis
The literature on pathogenesis is extensive and more than 100 years old. A search of the recent literature accessed by Entrez using the term viral pathogenesis pulls up more than 426,000 references, whereas a search of viral disease pulls up more than 695,000 references. Within this complex and voluminous literature, terms are used in multiple ways. Often the utilization of terms is predicated on historical factors rather than more up-to-date considerations. We will therefore begin by defining terms as they will be used in this chapter.
Productive, Abortive, and Latent Infection
Infection is the process by which a virus introduces its genome into a cell. Infection is productive if new infectious virus is made and abortive if no new infectious virus is produced. Infection is latent if the production of infectious virus does not occur immediately but the virus retains the potential to initiate productive infection at a later time. The process of reinitiating a productive infection cycle from the latent state is termed reactivation. Latency is not merely a slow productive replication cycle; latency represents a unique transcriptional and translational state where infectious virus is not present, but where a productive replication cycle can be reinitiated when the need arises. A cell is permissive if it can support productive infection and nonpermissive if infection cannot occur at all or is abortive.
Acute Versus Chronic or Persistent Infection
Acute infection occurs when a virus first infects a susceptible host (Fig. 10.1). Chronic or persistent infection is the continuation of infection beyond the time when the immune system might reasonably be expected to clear acute infection. The
terms chronic and persistent have been used interchangeably for many years; we will continue this convention. It is important to note that these terms denote the presence of viral infection in the host for long periods but do not provide insight as to the mechanism(s) responsible for prolonged survival of the virus in the host. Mechanisms responsible for chronic infection include persistence of nucleic acid, continuous replication, latency, and reactivation. More than one of these processes may occur at the same time. In some cases, viral nucleic acid can be detected in the host for prolonged periods, although the nature of the infectious process has not been defined (see Fig. 10.1). In such cases, chronic infection may represent continuous replication, latent infection, abortive infection without clearance of residual nucleic acid, or perhaps some as-yet-unidentified form of viral infection.
terms chronic and persistent have been used interchangeably for many years; we will continue this convention. It is important to note that these terms denote the presence of viral infection in the host for long periods but do not provide insight as to the mechanism(s) responsible for prolonged survival of the virus in the host. Mechanisms responsible for chronic infection include persistence of nucleic acid, continuous replication, latency, and reactivation. More than one of these processes may occur at the same time. In some cases, viral nucleic acid can be detected in the host for prolonged periods, although the nature of the infectious process has not been defined (see Fig. 10.1). In such cases, chronic infection may represent continuous replication, latent infection, abortive infection without clearance of residual nucleic acid, or perhaps some as-yet-unidentified form of viral infection.
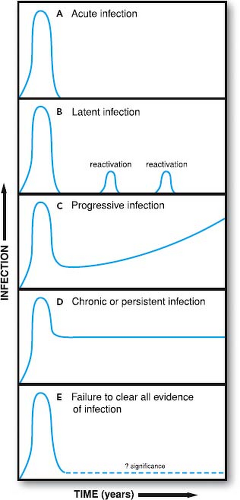 Figure 10.1. Schematic of different patterns of viral infection. |
In some cases, as for HBV or hepatitis C virus (HCV), a proportion of persons become chronically infected while others are cured. In these cases, the transition from acute to chronic is arbitrarily defined as the time when most patients have cleared acute infection. In other cases, essentially all hosts become chronically infected, as is seen with herpesviruses or lentiviruses such as human immunodeficiency virus (HIV). In this case, the transition between acute and chronic infection is defined as the time required for clearance of the initial burst of viral replication and establishment of equilibrium between the host and the virus.
There are two primary mechanisms for establishment of chronic infection: continuous replication and establishment of latency. During latent viral infection, the virus has a genomic and transcriptional strategy, often involving restricted viral gene expression, which allows the genome to survive even when lytic replication is not occurring. Examples include the proviral form of retroviruses or the circular episomal form with selective expression of viral genes observed for herpesviruses such as Epstein-Barr virus (EBV) and herpes simplex virus (HSV). Often, latently infected cells express no viral proteins, making latency immunologically silent. This is the ultimate form of immune evasion, as the host has no known mechanisms for sensing the presence of the virus. To survive and spread from the latently infected cell, the virus must be able reactivate and reinitiate the lytic cycle of gene expression, potentially generating antigens that the immune system can respond to.
To succeed via continuous replication, a virus must generate new infectious virions despite ongoing innate and adaptive immune responses. Even for viruses that utilize the establishment of latency as a primary strategy, intermittent reactivation and replication may be required to maintain latency and to spread from host to host. Some viruses, such as HIV, persist via both continuous replication and establishment of latency, presenting a particularly difficult challenge for the host immune system.107,301
Quasispecies
The mixture of viruses present in the host at a given time is a quasispecies. Although it is convenient to think of a virus as a single homogeneous agent, this is not true because both viral RNA and DNA polymerases make errors that generate mutant viruses during infection. The polymerases of RNA viruses are generally less accurate in copying template molecules than those of DNA viruses; mutation may therefore play a greater role in RNA than DNA virus pathogenesis. However, mutation may play a role in the pathogenesis of any virus. The quasispecies generated by mutation is acted on by complex and powerful selective pressures in the host, with some viruses having a survival or fitness advantage over others. The nature of these selective pressures and the pathogenic potential of the selected viruses are important determinants of viral pathogenesis.
Control of Acute Versus Chronic Infection
The distinctions between acute and chronic or persistent infection are very important. The viral genes and host immune
factors that foster or control acute versus chronic infection are distinct. For example, the cytokine interferon-γ (IFNγ) regulates latency and continuous replication of the murine γ HV68 (also referred to as MHV-68) but has at most a minimal effect during acute infection.16 This indicates that certain host responses are more relevant to chronic than acute infection. Control of either acute or chronic infection may involve responding to viral quasispecies. This is the case for both HIV and HCV infection, presenting the infected host with the overwhelming problem of dealing with many antigenically distinct viruses with different pathogenic strategies. Many viruses, including herpesviruses, papillomaviruses, and retroviruses, use different gene or gene expression patterns to succeed during acute versus chronic infection. For these reasons, it is fundamentally important not to consider chronic infection as a mere continuation of acute infection.
factors that foster or control acute versus chronic infection are distinct. For example, the cytokine interferon-γ (IFNγ) regulates latency and continuous replication of the murine γ HV68 (also referred to as MHV-68) but has at most a minimal effect during acute infection.16 This indicates that certain host responses are more relevant to chronic than acute infection. Control of either acute or chronic infection may involve responding to viral quasispecies. This is the case for both HIV and HCV infection, presenting the infected host with the overwhelming problem of dealing with many antigenically distinct viruses with different pathogenic strategies. Many viruses, including herpesviruses, papillomaviruses, and retroviruses, use different gene or gene expression patterns to succeed during acute versus chronic infection. For these reasons, it is fundamentally important not to consider chronic infection as a mere continuation of acute infection.
Equilibrium and Nonequilibrium States in Pathogenesis
A fundamental concept in pathogenesis is that acute infection is a nonequilibrium state, whereas chronic infection is a metastable equilibrium between virus and host. During acute infection, both the host response and virus infection change continuously until infection is resolved or progresses to death of the host or establishment of chronic infection. In contrast, chronic infection, once established, is an equilibrium process with viral and host processes balancing each other. In particular, the immune system of the host brings the acute infection under control and delays or prevents a chronic infection from killing the host. Progression of chronic infection to disease often reflects a change in this equilibrium (see Fig. 10.1). Even when the host and virus are in a stable equilibrium, the infectious process is dynamic with host and viral processes balanced on a knife-edge such that small changes in either virus or host can disrupt the equilibrium with devastating consequences.
An example of nonequilibrium and equilibrium states during acute and chronic infection is provided by EBV, a γ-herpesvirus that causes acute mononucleosis in humans. During the acute stage of infection, EBV causes rapid B-cell expansion (a nonequilibrium process) that is then brought under control by the expansion of virus-specific T cells, which act to maintain the virus in a latent state (an equilibrium process). When levels of latently infected B cells are assessed, significant individual-to-individual variation is observed but levels are remarkably constant within a given individual over time. However, when patients are immune suppressed, the level of latently infected B cells increases,12 suggesting that interactions between the virus-specific T cells and the virus maintain equilibrium during the chronic stage of infection. This balance between immune and viral processes during chronic infection is incompletely understood in most cases and yet is fundamentally important for understanding many human diseases.
Disease
Disease is a harmful pathologic consequence of infection. In many cases, infection is apparently harmless to the host and does not result in disease. One of the most important goals of pathogenesis research is to define in molecular terms what determines the difference between infection and disease. Even highly virulent viruses often establish infection in a greater number of hosts than they cause disease. Viruses such as rabies, Ebola, or HIV, which cause significant disease in nearly all infected persons, are the exception.
Disease may be associated with cell and tissue destruction (as in rabies virus killing neurons), induction or secretion of inflammatory cytokines (as in the induction of fever by many viruses), cellular dysfunction induced by viral infection (as in the case with lymphocytic choriomeningitis virus [LCMV] infection of the pituitary), paracrine effects of viral gene products (as in induction of angiogenesis by Kaposi’s sarcoma herpesvirus [KSHV]), and the induction of malignant tumors to the effects of the immune system as it responds to infection (as in immunopathology seen with many viruses) or to the presence of a specific virus interacting with allelic polymorphisms in the host to trigger disease.40,300
In many cases, virus-associated disease is defined as a series of nonspecific symptoms or signs such as fever, malaise, or anorexia. The presence of these symptoms and signs is common to infection with many different pathogens and therefore provides little insight into the mechanisms of viral pathogenesis. Recent work with microarray technology suggests that even the nonspecific syndromes associated with acute virus infection with different viruses may be distinguished by the pattern of host gene expression. It is likely that pathogenesis researchers will use such molecular signatures to distinguish between infections with different viruses and to define host genes involved in viral pathogenesis.
In contrast to the nonspecific syndromes commonly associated with virus infection, the presence of specific symptoms or signs of disease such as hepatitis, immunodeficiency, pocks on the skin, or paralysis provides important clues as to the nature of the pathogenic process. It is this relationship between the signs and symptoms of disease and the presence of infection with specific viruses that has led to the close link between clinical observations of the natural history of disease and the science of viral pathogenesis.
Virulence
Virulence—the relative capacity of a virus to cause disease—determines the relationship between infection and disease. Virulent viruses cause disease in a greater proportion of infected hosts, and cause more severe disease, than viruses of lower virulence. Virulence comes in many forms, from the induction of rapid death as for variola major (the causative agent in smallpox) to the induction of tumors over prolonged periods, as is the case with certain papillomaviruses or herpesviruses, to the induction of organ failure over many years, as is the case with chronic HBV or HCV infection. Thus, the manifestations of virulence highly depend on the strategies that a given virus uses during infection.
The determination of whether one virus (e.g., Ebola virus) is more virulent than another virus (e.g., papillomavirus) represents a qualitative judgment, because it involves determining whether virus-induced hemorrhagic fever (as is seen with Ebola virus) is worse than metastatic cancer (as is seen with papillomaviruses). To avoid biases inherent in comparing different types of disease, virulence is properly used to compare the disease-inducing capacity of related viruses, such as different strains of the same virus. For example, Ebola Reston, which is not associated with human disease, is less virulent in humans than Ebola Zaire.
It is commonly assumed that viruses that replicate efficiently in the host are more virulent than viruses that replicate less efficiently, and indeed replication is an important determinant of the severity of disease in many cases. However, virulence is much more complex than simply the efficacy of replication. Other aspects of pathogenesis, including tropism, the host response to infection, and interactions between the virus and host tissues, play key roles in viral virulence. Therefore, virulence reflects host resistance to infection functioning in counterpoint to viral virulence genes and determinants with the outcome for the host hanging in the balance.
Invasiveness
Invasiveness is the capacity of a virus to enter into and damage a tissue, a property that distinguishes viruses with high potential virulence but differ in the efficiency with which they enter target tissues. For example, a virus may be highly virulent if directly inoculated into the central nervous system (CNS) but unable to cause disease if inoculated into the periphery, whereas a related virus with a mutation allowing it to cross the blood–brain barrier into the CNS can cause lethal disease following either peripheral or intracranial inoculation. This concept is nicely exemplified by the identification of neuroinvasiveness determinants in Sindbis virus that confer the capacity to cause lethal encephalitis.66
Cell-Intrinsic Versus Cell-Extrinsic Mechanisms
Because infected cells live in a tissue in the host, they are affected both by events that occur inside the cell and by events that influence the cell from the outside. Events that occur in a cell independent of events outside of the infected cell are termed cell intrinsic. Some cell-intrinsic determinants of infection are owing to intrinsic cellular resistance to infection conferred by the presence of molecules that block viral infection. Events that are dictated by processes that occur outside of the cell are termed cell extrinsic. Many cell-extrinsic events are owing to innate and adaptive immunity. It is often the case that processes occurring in infected cells or tissues are affected by both cell-intrinsic and cell-extrinsic mechanisms. An example is the induction of death of infected cells that may be owing to cell-intrinsic induction of programmed cell death or cell-extrinsic immune factors such as cytokines or perforin and granzymes.
Evasion of Host Molecules and Mechanisms
Most viruses have evolved mechanisms to counter host innate and adaptive immunity or to bypass intrinsic cellular resistance molecules so that the virus can complete the infectious process and spread to a new host. These mechanisms constitute viral evasion of host responses. Often, evasion strategies involve viral genes with close homology to host genes, as when a virus encodes a host cytokine or cytokine receptor mimic. Other evasion strategies utilize molecules with novel structures to avoid host responses. Because the mechanisms responsible for acute and chronic infection differ, both with regard to viral and host factors, it follows that immune evasion mechanisms are different for acute versus chronic infection. During acute infection, viral immune evasion strategies commonly focus on the host innate immune response, whereas evasion of adaptive immunity is more important for maintaining chronic infection. When analyzing immune evasion mechanisms, it is therefore important to identify the aspects of the infectious process for which a given molecule is important.
Subversion of Host Molecules and Mechanisms
Viruses must be able to utilize host metabolic and regulatory systems to optimize their growth, survival, and spread. In some cases, the normal functions of the host cell may be sufficient for the virus to replicate. In other cases, host cellular processes may be insufficient or have deleterious effects on the virus. Subversion refers to the ability of a virus to redirect or alter normal host processes to the advantage of the virus. For example, some viruses subvert the function of chemokine receptors or other immune-signaling proteins to supply intracellular signals that regulate viral gene expression or encode molecules that subvert the cyclin-dependent progression of cell cycle. There are many different signaling pathways and cellular processes that are subverted by viruses; in fact, it is reasonable to presume that essentially every pathway and process within the cell is subverted by one virus or another. This is the inevitable consequence of the obligate intracellular lifestyle of viruses and their capacity to evolve more rapidly than the mammalian host.
Tropism
Tropism is the capacity of a virus to infect or damage specific cells, tissues, or species. It is a fundamentally important contributor to viral pathogenesis and virulence, as the capacity to induce disease depends on the cell and tissue infected. For example, a neurotropic virus such as West Nile Virus can cause encephalitis or paralysis, whereas a virus with tropism for CD4 T cells such as HIV causes immunodeficiency.
One key determinant of viral tropism is the cognate interaction between the viral cell attachment protein(s) and receptor(s) present on host cells. However, there are many additional factors that determine cell, tissue, and species tropism. The concept of tropism is rapidly evolving with the recognition that essentially every aspect of the viral infectious process within a cell or tissue can be a determinant of tropism. Both cell-intrinsic and cell-extrinsic factors may alter viral tropism.
Essential Genes, Virulence Genes, and Virulence Determinants
Virulence is determined by the capacity of a virus to grow, be invasive, infect vulnerable cells, evade the immune system, subvert cellular processes, and cause tissue damage. These capacities are encoded in the viral genome, by alleles of individual virulence genes. That viral genotype confers pathogenic capacity was proven by studies using reassortant viruses, mutant viruses, or molecularly cloned viruses to show that alterations in genotype confer virulence phenotype (e.g., 10,55,56,66,69,121,168,229,265,287,291,308). Subsequent studies have shown that different viral genes can confer capacities for infection in different tissues, indicating that viral virulence genes are tissue specific.109,175 Consistent with this, viral variants that arise during chronic infection are often cell and tissue specific.2,3,108,143,189
Any gene essential for replication contributes to virulence, because viruses must replicate to complete their life cycle. In this sense, all viral genes involved in replication are virulence genes. As this is not a very useful concept, viral genes essential for replication in permissive cells are termed essential genes rather than virulence genes. Virulence genes are not required for replication per se but are important for virulence in the
host. Although the distinction between an essential gene and a virulence gene is useful, it is important to recognize that allelic variations in the function of essential genes can be important determinants of viral virulence. In this case, the specific property associated with virulence is termed a virulence determinant. Thus, virulence determinants can be allelic variants of essential genes that confer significant differences in pathogenesis.
host. Although the distinction between an essential gene and a virulence gene is useful, it is important to recognize that allelic variations in the function of essential genes can be important determinants of viral virulence. In this case, the specific property associated with virulence is termed a virulence determinant. Thus, virulence determinants can be allelic variants of essential genes that confer significant differences in pathogenesis.
The size of the viral genome puts boundaries on the number of genes that a virus can use for pathogenesis and replication. For example, smaller RNA viruses often have genomes of less than 10,000 nucleotides, whereas poxviruses or herpesviruses may have genomes greater than 200,000 nucleotides in length. However, viral proteins often have multiple independent functions—a property particularly well documented for RNA viruses. Thus, the pathogenic capacity of a virus with a small genome should not be underestimated.
Virus particles can contain virulence determinants that are not encoded in their genomes. For example, vaccinia virus incorporates host complement regulatory proteins into its envelope, thereby becoming resistant to inactivation by host complement proteins.294 Another type of epigenetic mechanism is exemplified by herpesviruses such as human cytomegalovirus (HCMV) and HSV that can package host and viral messenger RNAs (mRNAs) into the virion.36,93,251,282 Although their role in pathogenesis is not yet clearly defined, carryover of these mRNAs can result in synthesis of viral proteins before the traditional transcription program of the virus is initiated.251,282
It is not necessarily true that viral virulence determinants exert their effects in host cells actually infected by the virus. One example of this is paracrine regulation of tissue pathology by virus-encoded cytokines. Another example is the movement of viral proteins from an infected to an uninfected cell. For example, the HSV protein VP22 can efficiently move from an infected cell to the nucleus of uninfected cells,68 can carry other proteins between cells,224 and can transport mRNAs from one cell to the next.251 This is a fascinating example of how a viral virulence determinant can act on uninfected cells to contribute to viral pathogenesis.
Virulence Genes and Determinants May Not Encode Proteins
Virulence can be conferred by viral promoters or other cis elements in the viral genome or noncoding RNAs. For example, EBV infection is associated with the development of B-cell lymphomas. The EBV EBNA1 protein plays an important role in the maintenance of latent EBV infection in B lymphocytes. EBNA1 can be expressed from any of four promoters—Cp, Wp, Fp, and Qp—each of which is regulated in distinct ways by viral and host proteins,264 ensuring that EBNA1 is expressed at the appropriate place and time in B cells. These finely tuned promoters are therefore properly considered virulence determinants for EBV-associated malignancies. Another example of non–protein-coding virulence determinants are virally encoded microRNAs, which are expressed by a number of mammalian viruses, including herpesviruses, adenoviruses, and polyomaviruses.228,257 These noncoding RNAs target multiple cellular processes, including cell cycle regulators, apoptotic machinery, or virus-encoded transcript, and play a role in promoting viral persistence. Noncoding elements also play important roles in the pathogenesis of several RNA viruses. For example, attenuating mutations within the internal ribosomal entry site (IRES) of the Sabin vaccine strain of poliovirus are important for replication in neurons and neurovirulence in mice and primates.97,99,141,149 It is becoming increasingly clear that the transcriptional complexity of a broad range of organisms is considerably higher than previously believed,23,117 and it is now recognized that this is also the case for viruses such as herpesviruses.46,47,133 It is likely that virulence genes that do not encode proteins will increasingly be recognized in many viruses.
Conceptualizing Viral Pathogenesis
Every fact or principle presented in this textbook relates in some way to the pathogenesis of infection or disease. Thus, it may seem that pathogenesis is too complex to be studied in any mechanistic detail, or that viruses differ so significantly that common principles do not exist. This problem is further compounded because many events and processes are going on at the same time in vivo. Furthermore, processes that occur in vivo are nonlinear (e.g., unrestricted viral replication may be exponential) or even stochastic (e.g., whether a mutation occurs when a genome is replicated), or otherwise are the results of as-yet-unidentified allelic variations in host genes that influence pathogenesis.1,300
Given this complexity, it is worth considering how the questions of pathogenesis are formulated and placed in context for interpretation. There are many ways to look at infection—three of which are presented here as a basis for understanding viral pathogenesis. Pathogenesis may be conceptualized as an organized process consisting of sequential stages, as the result of stochastic events under selection by bottlenecks in infection, or as the integration of host allelic variations in genes that determine resistance to infection. Each view of pathogenesis has its limitations and benefits as a basis for understanding the nature and mechanisms of viral infection.
Clinical Observations Define Fundamental Pathogenesis Questions
Viral pathogenesis has been studied for longer than any other aspect of virology; however, in many ways, we know less about pathogenesis than we do about other aspects of virology. The pathogenesis of viral disease was studied before viruses were even identified. Smallpox was known before variola virus was identified, and rabies virus was propagated in animals and an effective vaccine was developed by Pasteur before the virus was characterized.131 It was known that chickenpox infection conferred lifelong resistance to disease before varicella-zoster virus (VZV) was identified. A study of measles in 1886 in the Faroe Islands identified the incubation period between exposure and disease, resistance of previously exposed individuals to reinfection, and the susceptibility of the very young to disease.95,96 These clinical observations often identify the questions that must be answered in pathogenesis studies. For example, mosquito-borne alphaviruses such as chikungunya virus cause severe acute and persistent arthralgia in infected humans, raising the question of how these viruses cause joint pain and whether chronic disease is associated with viral persistence. Through the careful analysis of human clinical samples and the use of appropriate animal models, the viral pathogenesis researcher can begin to define the molecular mechanisms underlying a disease process.
Conceptualizing Viral Pathogenesis as a Series of Sequential Stages in Infection
A very useful way to conceptualize viral pathogenesis is as a series of sequential stages resulting in survival and spread of a virus through the infected host and on to a new host (Figs. 10.2 and 10.3). Different viruses may utilize distinct molecular mechanisms to accomplish each of these stages. For example, all viruses must infect cells on entry into the host and must spread to new hosts. As the host has mechanisms to inhibit each of these stages of infection, the stages of infection may be considered as hurdles over which the virus must jump to survive.
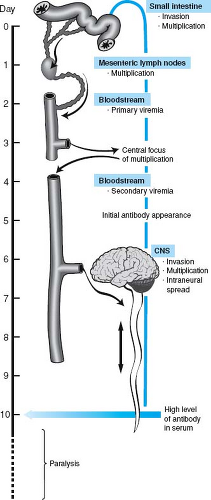 Figure 10.2. Model of poliovirus pathogenesis as a series of sequential stages. The steps in poliovirus infection of a human are schematized. |
There may be more than one way for a virus to overcome such a hurdle; however, different viruses may utilize the same strategy to overcome a given hurdle. An example of a common strategy is the relative resistance of some viruses to inactivation in water or sewage, allowing them to spread between hosts by fecal–oral transmission. Another example is infection of the thymus resulting in deletion of virus-specific T cells as self-antigens. Yet another common strategy is evading immunity via the establishment of latent infection. Thus, there are many common strategies for infection shared by different viruses.
Dividing viral pathogenesis into stages is a very useful way to conceptualize the infectious process but has significant limitations. First, dividing pathogenesis into stages creates the impression that events are both sequential and depend on each other. This is not always the case; many independent events may be going on at the same time in the host. Second, this conceptual framework suggests that completion of a stage is associated with initiation of the next stage; a significant oversimplification. For example, viremia may continue after a virus has entered the CNS to cause encephalitis. The utility of considering viral pathogenesis as a series of sequential events must be balanced against how well this conceptual framework fits actual events during infection.
Poliovirus pathogenesis provides an excellent example of how pathogenesis can be broken down into a series of steps that culminate in either virus-induced disease or viral control. Clinical questions that drove poliovirus pathogenesis research through the first half of the past century included why poliomyelitis emerged in the 20th century, what mechanisms were responsible for paralysis, and how did protective immunity work.183,234,235,274 Solving these questions was central to combating a very important and pressing public health problem. Infection with poliovirus in humans has a wide range of possible outcomes from asymptomatic infection to meningoencephalitis with or without paralysis.183,234,235,274
Studies over many years identified and analyzed a series of stages of poliovirus infection, leading to a relatively simple model for the pathogenesis of disease that elegantly explains paralysis, the low proportion of infected hosts paralyzed, and the lifelong immunity conferred by prior infection. This model, one of the most useful ever constructed, provided a basis for developing the poliovirus vaccines that have largely, although not completely, eliminated paralytic poliomyelitis as a scourge of humanity.
The stages in poliovirus pathogenesis according to this model are outlined in Figure 10.2.24,183,234,235 The virus enters the intestine via the fecal–oral route, binds to M cells overlying the Peyer’s patch, is transported into the intestinal wall, and then replicates in lymphoid cells, leading to a primary viremia and infection of secondary sites. Replication in secondary sites gives rise to a secondary viremia that reaches a level capable of initiating CNS infection.234,235 CNS infection involves passage of the virus across the blood–brain barrier to infect neurons within the CNS. The blood–brain barrier is viewed as an
important anatomic barrier to infection of the CNS, and passage across this barrier is poorly understood. Alternatively, the virus may spread via the blood to peripheral nerves and then spread up the nerves to enter the CNS.98,185,205,235,241 Within the CNS, the virus infects motor neurons; destruction of these cells leads to paralysis. Certain motor neurons are hypothesized to more susceptible to poliovirus infection than others, and some poliovirus strains are either more invasive or more likely to kill neurons than others; these variables contribute to variation in disease penetrance and severity.
important anatomic barrier to infection of the CNS, and passage across this barrier is poorly understood. Alternatively, the virus may spread via the blood to peripheral nerves and then spread up the nerves to enter the CNS.98,185,205,235,241 Within the CNS, the virus infects motor neurons; destruction of these cells leads to paralysis. Certain motor neurons are hypothesized to more susceptible to poliovirus infection than others, and some poliovirus strains are either more invasive or more likely to kill neurons than others; these variables contribute to variation in disease penetrance and severity.
 Figure 10.3. Model of poxvirus pathogenesis as a series of sequential stages. The steps in ectromelia virus pathogenesis are schematized. The outcome of ectromelia infection varies with mouse and viral strain.39,75 The viremia is thought to be cell associated. The histopathologic image of a pock was provided by Drs. Mark Martinez and Peter Jahrling and is skin from a variola-infected monkey stained with specific antisera to identify the infected cells shown as poxvirus antigen in the inset image. (Adapted from Fenner F, Buller RM. Mousepox. In: Nathanson N, Ahmed R, Gonzalez-Scarano F, et al, eds. Viral Pathogenesis. Philadelphia: Lippincott-Raven, 1997:535–553.) |
Concurrent with entry into the lymphatic system, an immune response is generated (see Fig. 10.2). It is hypothesized in this model that immune antibody limits access to the CNS and prevents paralytic disease.183,235 Antibody might act by preventing virus in the circulation from crossing the blood–brain barrier and entering the CNS. However, antibody is capable of inhibiting neural spread of viruses and can inhibit viral infection by acting directly on or within neurons.160,183,290,292,299 Regardless of the mechanisms by which antibody protects, the outcome of infection is a race between the virus and the immune system, presenting another explanation for variations in clinical outcome. The immune system wins if antibody is made early enough to prevent spread to the CNS and neuronal destruction. The virus wins if infection of motor neurons occurs prior to development of protective antibody responses.
This model provides a basis for understanding many aspects of poliovirus infection, disease, immunity, and vaccination; however, many questions are left open. Questions that remain unanswered at the molecular level include how the virus enters the body, the molecular basis of cell and tissue tropism, the mechanism by which antibody acts to prevent CNS disease, the mechanisms of induction of immunity, and the route(s) by which the virus spreads.
Importantly, although the mechanistic details underpinning the pathogenesis of different viruses will differ significantly, the broad stages of the systemic infection process outlined for poliovirus pathogenesis are also relevant for a wide array of other viral pathogens. For example, although poxviruses are very different from small RNA viruses, such as poliovirus, there are remarkable parallels between the models of poliovirus infection (see Fig. 10.2) and those for poxviruses (Fig. 10.3). This includes the concept that both viruses must reach a threshold of viremia to facilitate viral invasion into target organs and that the disease process represents a race between the virus and the host immune response, where the outcome of that race plays a major role in determining whether the virus is controlled or if disease results. Refer to the eBook for a more complete description of poxvirus pathogenesis as a stepwise process. These general principles extend to a wide array of viral pathogens, including arboviruses, herpesviruses, and lentiviruses.
Conceptualizing Viral Pathogenesis as the Interaction Between Stochastic Events and Bottlenecks in Infection
It is important to recognize that the process of infection has, in addition to a series of predictable events that occur in sequence, an element of stochastic variation that contributes to real variability to the outcome of infection. Thus, viral pathogenesis can be conceptualized as a series of stochastic events with the process of infection determined by strong selective pressures in the host referred to as bottlenecks in infection (Fig. 10.4). These bottlenecks can be thought of as analogous to the rate-limiting step in a chemical reaction. Stochastic events such as mutations or the random success of a given virus at a stage in infection provide variation in the substrates for these rate-limiting steps.
The concept that random events contribute to pathogenesis is not a comfortable one for investigators. However, even using clonal virus stocks for inoculation of genetically identical mice of the same sex and age results in viral titers in tissues that vary considerably (even by orders of magnitude) from one mouse to the next. This variation is not experimental error or noise but is rather a real phenomenon relevant to understanding infection. During natural infection of genetically variable hosts with viral quasispecies, the variation is likely to be much larger.297
The reasons for the occurrence of significant variations even when conditions of infection are apparently homogeneous are not clear but likely involve several different factors, including epigenetic changes within the host, specific interactions between viruses and certain host genes that confer disease,44,300 environmental variables, and viral factors such as viral mutation rates that drive the generation of mutants with selective advantages in the host. This latter process, which involves the random generation of mutant viruses—a subset of which may provide a selective advantage that allow the virus to overcome bottlenecks and efficiently spread throughout the host—represents an important component of viral pathogenesis. For example, polioviruses that have high-fidelity polymerases, and therefore are less able to generate mutant viruses, are less efficient in infecting target tissues and causing disease, suggesting that the generation of viral quasispecies allows the virus to overcome bottlenecks and is essential for disease pathogenesis.296
Selective pressures at bottlenecks in infection can select mutants from within the viral quasispecies that have a fitness advantage (see Fig. 10.4). This is illustrated experimentally by studies with Venezuelan equine encephalitis virus (VEE), an equine pathogen that infects both humans and mice. Following subcutaneous inoculation, VEE initially replicates in the draining lymph node before seeding a serum viremia that leads to viral dissemination and ultimately viral entry into the CNS. Studies that compared molecularly clones of wild-type VEE to a mutant virus with a defined mutation at position 76 in the E2 envelope protein demonstrated that the mutant virus replicated within the draining lymph node but failed to spread systemically. Importantly, infection with the mutant virus led to the rapid generation of viruses that were able to efficiently spread within the host, and characterization of these viruses found that some contained a reversion of the original E2 mutation to wild-type. However, other viruses maintained the original E2 mutation but developed second site mutations that conferred the ability to disseminate systemically.10,94 These results demonstrate that there may be many ways to overcome
a specific bottleneck. Bottlenecks may also be responsible for significant variations in the kinetics of infection between hosts. For example, a variant capable of bypassing a given bottleneck may arise sooner in one host than in another. Refer to the eBook for an expanded discussion of how VEE rapidly mutates to overcome bottlenecks in infection.
a specific bottleneck. Bottlenecks may also be responsible for significant variations in the kinetics of infection between hosts. For example, a variant capable of bypassing a given bottleneck may arise sooner in one host than in another. Refer to the eBook for an expanded discussion of how VEE rapidly mutates to overcome bottlenecks in infection.
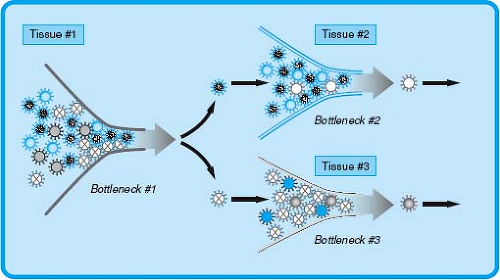 Figure 10.4. Viral pathogenenesis as stochastic events followed by selection by bottlenecks in infection. Shown is a representation of a viral quasispecies, only two of which are fit to pass through an initial bottleneck in infection. Subsequent replication of these viruses in different tissues generates further quasispecies that are again acted in by selective forces at further bottlenecks in infection. |
An example of a bottleneck in infection exerting a selective pressure comes from HIV infection. When an infected person exposes an uninfected person to the diverse HIV quasispecies that develops during chronic infection, the viruses that emerge in the new host represents only a subset of the viruses in the inoculum.106,301 This suggests the existence of a bottleneck early in infection through which some viruses selectively pass. Detailed study shows that HIV viruses that utilize the CCR5 co-receptor are more efficient at spread and initial replication in both men and women than viruses that utilize CXCR4 as a co-receptor.167,214,231 This suggests that co-receptor utilization is a critical event for passing through a bottleneck during the initial stages of infection. After passage through this bottleneck, HIV isolates that utilize CCR5 are less fit for later events in infection leading to selection of viruses utilizing CXCR4 as a co-receptor. Thus, a virus that is more effective at passing one bottleneck in infection can be replaced, via mutation and selection, by viruses more fit for bypassing subsequent bottlenecks in infection. For these reasons, one must not assume that the virus initially entering the host is the virus responsible for viral disease but must consider stochastic events and selective pressures in models of pathogenesis.
An important consequence of passage through a bottleneck is contraction of quasispecies heterogeneity. When a mutant virus is selected, allelic variations in regions of the genome that are not under selective pressure are carried along through the bottleneck. Thus, random mutation followed by selection can generate novel viruses with properties that are not selected for at all and may therefore have unique pathogenic properties. For example, a virus may mutate sequences encoding a T-cell epitope to escape control by T lymphocytes, and this new sequence may confer previously unexpected properties to the virus. Therefore, selective pressure may result in the emergence of different viruses in different hosts, providing a mechanism for the emergence of viruses with new properties in a population of susceptible individuals.
Conceptualizing Viral Pathogenesis as the Integrated Effects of Host Genetic Variation
The previously discussed ways to conceptualize viral pathogenesis are highly focused on the virus, but it has been clear since the earliest studies of viral pathogenesis that hosts differ significantly in genetic susceptibility to infection.44,301 The major host determinant of viral virulence and pathogenesis is innate and adaptive immunity, but host genes not involved immunity also play a role. Allelic variations in these host genes can alter viral pathogenesis (Fig. 10.5).
The extent to which allelic variations in host genes control viral pathogenesis is only now being appreciated. Some examples are well established, whereas others come from single studies and await confirmation. Mutations in CCR5 confer resistance to HIV infection.57,164,248 Human noroviruses (type virus Norwalk) are responsible for more than 90% of the epidemic nonbacterial gastroenteritis in the world.61,90 Norwalk virus susceptibility is determined by blood group secretor status conferred by the presence of the FUT2 fucosyltransferase.127,163 Among human norovirus strains, there are multiple patterns of virus-like particle (VLP) binding to blood group carbohydrates, suggesting that allelic variation in human blood groups contribute to susceptibility to a variety of norovirus strains. Patients with mutations in the IFNγ receptor have been reported to have unusual viral syndromes.226 Autosomal dominant mutations in the chemokine receptor CXCR4 have been associated with severe warts,154 and mutations in EVER1 and EVER2 have been associated with an unusual clinical presentation of papillomavirus infection called epidermodysplasia verruciformis.237 Allelic variations in mannose-binding lectin and FcγRIIA have been linked to the severity of severe acute respiratory syndrome (SARS).130,320 A relationship between expression of certain KIR genes, encoding NK cell receptors, and severity and chronicity of infection with HIV, HCV, and EBV have been reported.43 Responsiveness to hepatitis B vaccine may be linked to allelic variations in complement genes.25,119 Thus, a significant number of allelic variations in
humans are important determinants of viral pathogenesis, and additional candidates for such variation are being found at a high rate. Refer to the eBook for further discussion of how variation in blood group secretor status affects susceptibility to human norovirus infection.
humans are important determinants of viral pathogenesis, and additional candidates for such variation are being found at a high rate. Refer to the eBook for further discussion of how variation in blood group secretor status affects susceptibility to human norovirus infection.
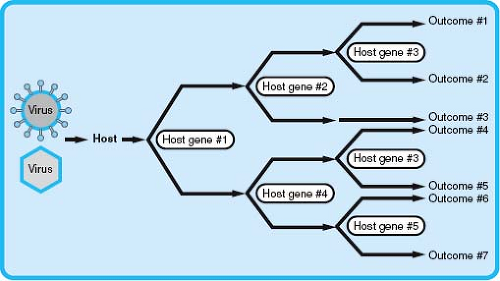 Figure 10.5. Viral pathogenesis as the result of the action of a series of host genes. Shown is a representation of infection with different viruses progressing through the host as acted on by host genes that confer either resistance or susceptibility to infection. Depending on the effects of host gene 1, one of two different patterns of infection emerges. In turn, additional host genes interact with the virus, resulting in different outcomes for the host. Note that the more host genes play a determining role, the greater the number of possible outcomes. |
Although many studies have focused on the impact of variation in a single gene on viral susceptibility, it is clear that susceptibility or resistance to viral infections is a complex phenotype regulated by multiple interacting genes and gene networks. For example, studies designed to identify genes that regulate resistance to influenza infection between two mouse strains have identified multiple quantitative trait loci (QTL) that contribute to disease susceptibility.28 Although the full number of host genes that vary between susceptible and resistant hosts for a single virus is only beginning to be understood, it is likely that variation in hundreds of different genes may contribute to disease resistance or susceptibility.44,302 Recent advances in the area of host genetics, including the application of genome-wide association studies (GWAS), which scan the genome for allelic variants, such as single nucleotide polymorphisms (SNPs), associated with specific phenotypes, as well as whole exome and whole genome sequencing, promise to further enhance our understanding of how different polymorphic genes contribute to disease susceptibility. For example, recent application of GWAS approaches to identify genes that associate with HIV control identified a major association between polymorphisms in the human leukocyte antigen (HLA) locus that affect class I peptide presentation to CD8 T cells and an individual’s ability to control HIV.221 These approaches have also been extended to HCV and recently identified a polymorphism within the IL28B gene (interferon λ3) that associates with viral clearance following therapy with interferon.84 These types of findings have direct implications for predicting treatment outcomes for infection in patient populations, as has been suggested for inflammatory diseases.300
The examples outlined previously illustrate that allelic variants in host genes can have a significant impact on the pathogenesis of infection (see Fig. 10.5). As multiple independently segregating genes contribute to the susceptibility or resistance to a viral pathogen, many human genotypes exist that may vary significantly in their resistance to even a single viral infection. Therefore, even without considering the impact of viral virulence determinants or other environmental factors, much of the variation in viral pathogenesis within a diverse population could be explained by polymorphisms in host genes. This has implications not only for viral pathogenesis in human populations but also for studying viral pathogenesis in animal models. Although GWAS and candidate gene studies in humans can establish an association between a polymorphic gene and susceptibility to a viral infection, the investigation of the mechanisms by which a polymorphic gene or genes affects pathogenesis requires additional tools, including appropriate animal models. However, pathogenesis studies are often performed in genetically homogeneous experimental hosts (e.g., inbred mouse strains), which results in the loss of any significant effect of host genetic variation on viral pathogenesis and limits investigation of how allelic variants affect pathogenesis outcomes. New model systems that more accurately model the genetic diversity that is present in human populations, such as the Collaborative Cross mouse RI panel,11 as well as classical mouse genetics systems, including BXD recombinant inbred lines,28 should enhance studies of the role of host variation and gene:gene interactions in viral pathogenesis.
The Study of Viral Pathogenesis
Many different tools must be used to understand the complex process of viral pathogenesis. Some of these tools have played essential roles in understanding pathogenesis and will continue to be useful. Examples include assays for infectious virus and histopathologic evaluation of infected tissues. Others are more recently developed and have only been applied in limited circumstances. Understanding the tools of experimental pathogenesis research is essential to understanding the interpretation of experiments that have contributed to our current understanding of virus infection.
Epidemiology
Epidemiology is an essential tool for pathogenesis research for defining patterns of disease and infection and the mode of transmission between hosts. Together with assays for prior infection such as serology or molecular detection of chronic virus infection, epidemiology can define the relationship between infection, immunity, and disease. Epidemiologic studies link a virus to a specific disease and allow formulation of the fundamental questions that must be answered to understand viral pathogenesis. This is nicely illustrated by the identification of Kaposi’s sarcoma (KS) herpesvirus, where epidemiology studies suggested that HIV status alone was not an accurate predictor of KS risk, indicating that an additional co-factor was responsible for KS.48,250 Following the discovery of KSHV,48 additional epidemiologic investigations convincingly linked KSHV infection with KS via demonstration that KSHV sequences were present almost universally in KS lesions and that seroconversion to KSHV preceded the development of KS.174
In some cases, epidemiologic investigation finds that known viruses explain but a part of the disease burden, leaving open the question of whether unknown agents contribute to disease. The process of associating new viruses with both viral and nonviral diseases continues to this day, with new viruses constantly emerging or being identified as responsible for already described diseases.
Animal Models
Animal models are fundamentally important tools for the study of viral pathogenesis. The need to understand human infection, combined with the complexities of doing human studies, has led to the use of animal models to ask pathogenesis questions that cannot be effectively answered via human studies. There are two types of animal models for human viral disease. In the first, one studies a human virus in infected animals. In the second, one studies an animal virus that is related to a human virus in its animal host. There is an essential tension between these two approaches; in one the “real” pathogen is studied, and in the other a “natural” infection is studied. In truth, each has its advantages and each its limitations.
Study of Human Viruses in Animal Models
Human viruses can be studied in animals that are susceptible to infection either because the virus does not exhibit species tropism or because tropism restrictions are overcome via genetic
manipulation of the host or virus. An important caveat of such studies is that human viruses seldom or never behave in an experimental animal exactly as they do in humans. Nevertheless, this is a very valuable approach. Examples include the use of mice to study HSV, Sindbis virus, yellow fever virus, VEE, LCMV, or chikungunya viruses; the use of humanized mouse models to study HCV and HIV; and the use of primates to study poliovirus, variola, HIV, and Ebola virus.
manipulation of the host or virus. An important caveat of such studies is that human viruses seldom or never behave in an experimental animal exactly as they do in humans. Nevertheless, this is a very valuable approach. Examples include the use of mice to study HSV, Sindbis virus, yellow fever virus, VEE, LCMV, or chikungunya viruses; the use of humanized mouse models to study HCV and HIV; and the use of primates to study poliovirus, variola, HIV, and Ebola virus.
Excellent examples of this approach are the analysis of infection with filoviruses such as Ebola or Marburg that are very difficult to study in infected patients. However, these viruses cause disease in macaques with significant similarities to human disease, including a striking hemorrhagic diathesis including disseminated intravascular coagulation.85,86,87 These animal models have been used to demonstrate that it is possible to vaccinate against filovirus infection136,270,319 and that passive transfer of antibody can be partially protective.105,216
Not all human viruses can replicate in animals. Five approaches have been taken to overcome this hurdle. These approaches are, first, passage-based adaptation of the human virus to growth in an experimental animal; second, engineering of the host to accommodate all or part of the pathogenesis of the human infection; third, expressing the virus as a transgene in an experimental animal; fourth, the creation of humanized mice where immunodeficient mice are reconstituted with aspects of the human immune system and components of the human target organ (e.g., the liver for HCV); and fifth, targeted modification of viruses to allow replication in a model host. Increasing knowledge of the mechanisms of viral pathogenesis and immunity holds out great hope for generations of better animal models.
In the first approach, a human virus is adapted to growth in an animal model. Ebola has adapted to infect guinea pigs and mice.35,52,89 Ebola infection of small animals is similar to primate and human Ebola infections in some ways. For example, dendritic cells (DCs) and monocytes are early targets of infection in all of the different models. However, mice and guinea pigs do not show the hemorrhagic diathesis seen in humans and macaques, which is a significant limitation for pathogenesis studies.35,52,85,89
In the second approach, the host is genetically engineered to allow analysis of a human virus. For example, transgenic expression of the poliovirus or measles virus receptors in mice confers susceptibility to intracerebral infection with poliovirus or measles virus.
In the third approach, the virus is expressed as a transgene in a live animal, allowing the replication cycle of the virus to proceed in certain cells even though the host is nonpermissive for infection. This has been accomplished for HBV with mice engineered to generate infectious virus from a transgenic viral genome.103,104
In the fourth approach, mice are used as hosts for human tissue allografts that can then be infected with human viruses. This approach is particularly useful for viruses that fail to replicate in nonhuman systems and has been applied to viruses such as HIV155 and VZV.14,249,321
In the fifth approach, the virus is manipulated in a specific way to allow infection of the animal to be used as a model. For example, based on an intimate knowledge of the mechanisms of lentivirus species tropism, it has been possible via manipulation of the HIV vif gene to create an HIV isolate that can replicate in macaques.113
Study of Animal Virus Infections That Resemble Human Infection
It is often impossible to approximate all aspects of the pathogenesis of a human virus in an animal model. An important alternative approach is to study the pathogenesis of an animal virus that is related to a human virus in an animal host. Examples include the study of murine cytomegalovirus (MCMV) as a model for HCMV, simian immunodeficiency virus (SIV) as a model for HIV, murine γHV68 as a model for human γ-herpesvirus infection, myxoma virus and ectromelia virus as models for smallpox, and murine norovirus infection as a model for human noroviruses. These models provide, especially in the murine system, tools for doing pathogenesis and host genetics that are unavailable in any other model.
An important caveat of this approach is that animal viruses seldom faithfully reproduce in every detail of a human disease. This is not surprising; after all, animals are not humans and animal viruses are not identical to human viruses. These dissimilarities are often used to argue against the utility of such animal models. However, many principles and mechanisms of viral pathogenesis have first been described in animal models and later proven relevant in humans.
Viral Genetics as a Tool for Analysis of Pathogenesis
Studies taking advantage of genetic differences between viral strains are as old as the study of pathogenesis itself. Comparisons between viral strains can identify correlates of virulence or attenuation but do not necessarily shed light on the mechanisms responsible for the phenomena observed. Reliance on naturally occurring strains of virus has strict limitations for defining pathogenesis mechanisms, because it is only by luck that genetic variation occurs between strains in a manner that allows a specific hypothesis to be tested. Thus, advent of sequencing, structural biology, and directed mutagenesis of viral genomes as tools for pathogenesis research has been a turning point for pathogenesis as an experimental science. Infectious clone technology now exists for most virus families, and by applying directed mutagenesis strategies to these molecularly cloned viruses, mechanistic hypotheses can be tested directly. Often, this is done using a loss-of-function genetic approach in which an entire viral gene is deleted or in which a mutation is used to ablate a specific biochemical function of a viral protein. By evaluating such viruses, one can link structural and biochemical properties of a protein to specific aspects of pathogenesis, thus making loss-of-function genetic approaches a fundamental part of current pathogenesis research.
It is important to understand the limitations of viral mutagenesis as an approach. First, loss-of-function genetic analysis depends on whether a viral property can truly be attributed to a specific mutation. Further, it is necessary to prove that a phenotype is not an artifact of changes that occur during manipulation of the viral genome. This is often accomplished by marker-rescue, in which the mutant virus is repaired to wild-type status and the resulting virus characterized. If this marker-rescued virus is different from the wild-type virus, it indicates that other mutations have occurred during the process
of mutant generation, thereby invalidating conclusions drawn using the mutant. This can be particularly problematic for rapidly evolving RNA viruses that may select compensatory mutations during generation of viral stocks. An alternative is to fully sequence the viral mutant—an approach that is now increasingly practical for even the largest viruses.
of mutant generation, thereby invalidating conclusions drawn using the mutant. This can be particularly problematic for rapidly evolving RNA viruses that may select compensatory mutations during generation of viral stocks. An alternative is to fully sequence the viral mutant—an approach that is now increasingly practical for even the largest viruses.
It is also necessary to prove that a phenotype is owing to a change in a viral protein and not to alterations in cis-elements in the viral genome or in adjacent genes that may confer phenotypes. For example, mutating a residue in a protein might alter a promoter for another viral gene or change the processing of a viral polyprotein. These problems are addressed by studying multiple distinct mutations in a protein, analysis of the expression of other viral proteins, or, when possible, complementing the mutation by expressing the protein in trans from another location in the virus.
A second limitation of loss-of-function genetics is that linking a mutation in a virus to a specific alteration in pathogenesis shows that the gene in question is necessary but does not indicate that the gene is sufficient for the virus to perform a specific task. For example, several genes might be important for an aspect of pathogenesis such that mutation of any one of them would give a phenotype; however, no single gene is independently sufficient for the virus to perform a certain task in vivo.
A third limitation of loss-of-function genetics is that the identification of an important role for a gene at a given step in pathogenesis does not provide information on the mechanisms responsible for the phenotype observed. In the absence of additional studies, one can only make correlative statements about the relationship between the behavior of a mutant virus and the biochemical properties of the protein encoded by the altered gene. For example, if a certain amino acid is important for binding of a host protein by a viral protein, and that same mutation causes the virus to be defective in pathogenesis, it is plausible that the phenotype observed in vivo relates to the binding between the host and viral protein. Proof of mechanism in vivo requires additional studies.
Interactions Between Host and Viral Genes in the Study of Pathogenesis
Defining the mechanism of action of a viral protein or gene in vivo is much more complex than showing that the gene is important, requiring that one determine experimentally why pathogenesis is altered by a specific mutation. It is this latter level of analysis that provides the greatest challenges. The attribution of changes in pathogenesis to a specific biochemical property of either the host or the virus is a complex task. The most important question is whether a change in the behavior of a virus in vivo is attributable to direct effects of a given mutation or is owing to indirect effects of the mutation. To exemplify this problem, consider the case of a mutant virus that both grows more poorly and induces a lesser inflammatory response than the wild-type virus. One interpretation of such data might be that the mutation influences the function of a gene that evades or subverts the host inflammatory response. A more trivial interpretation is that the host responds to a virus that grows poorly with a lesser inflammatory response. In this situation, one cannot cleanly attribute an immune evasion property to the viral protein in the absence of additional data.
This problem is conceptually similar to the Heisenberg uncertainty principle in physics in which detecting an electron requires the use of a particle that in turn changes the location of the electron. Analogously, one determines the function of the viral gene by mutating the gene; however, mutating the gene can change the process of infection sufficiently to complicate interpretation of any differences observed in pathogenesis.297 A major challenge in defining mechanisms of viral pathogenesis is then to identify genetic, structural, and pathogenesis approaches that allow attribution of events in vivo to specific biochemical mechanisms.
One method for doing this involves using a combination of host and viral genetics in a process termed host complementation. For example, the ICP34.5 gene of HSV is essential for viral virulence.27,50 One activity of ICP34.5 is to bind protein phosphatase 1A (PP1A) and to redirect PP1A phosphatase activity to dephosphorylation of eIF2α, thereby reversing the eIF2α-dependent antiviral effects of PKR.51,115,116,245,277 If ICP34.5 is acting to promote virulence through interactions with PKR, the ICP34.5 mutant should exhibit restored virulence specifically in mice lacking PKR, but not mice lacking other antiviral effector molecules. In fact, deletion of PKR, but not RNAse L, from mice fully restores the virulence of an ICP34.5 mutant virus,156 thereby establishing that ICP34.5 interacts with PKR in vivo. Other examples of this approach include the role of the influenza virus NS1 gene in countering STAT-1 dependent innate immunity,83 the role of the murine γHV68 complement regulatory protein v-RCA in evasion of complement responses,138 and the role of the m157 gene of MCMV as natural killer (NK) cell–activating receptor ligand.9,78,260 In all of these studies, deletion of a host gene or cell type has been used to define the mechanisms of action of a viral gene during infection.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


