31
Opioid Agonists & Antagonists*
CASE STUDY
A 60-year-old man with a history of moderate chronic obstructive pulmonary disease presents in the emergency department with a broken hip suffered in an automobile accident. He complains of severe pain. What is the most appropriate immediate treatment for his pain? Are any special precautions needed?
Morphine, the prototypic opioid agonist, has long been known to relieve severe pain with remarkable efficacy. The opium poppy is the source of crude opium from which Sertürner in 1803 isolated morphine, the pure alkaloid, naming it after Morpheus, the Greek god of dreams. It remains the standard against which all drugs that have strong analgesic action are compared. These drugs are collectively known as opioids and include not only the natural and semisynthetic alkaloid derivatives from opium but also synthetic surrogates, other opioid-like drugs whose actions are blocked by the nonselective antagonist naloxone, plus several endogenous peptides that interact with the different subtypes of opioid receptors.
 BASIC PHARMACOLOGY OF THE OPIOIDS
BASIC PHARMACOLOGY OF THE OPIOIDS
Source
Opium, the source of morphine, is obtained from the poppy, Papaver somniferum and P album. After incision, the poppy seed pod exudes a white substance that turns into a brown gum that is crude opium. Opium contains many alkaloids, the principal one being morphine, which is present in a concentration of about 10%. Codeine is synthesized commercially from morphine.
Classification & Chemistry
The term opioid describes all compounds that work at opioid receptors. The term opiate specifically describes the naturally occurring alkaloids: morphine, codeine, thebaine, and papaverine. In contrast, narcotic was originally used to describe sleep-inducing medications, but in the United States, its usage has shifted into a legal term.
Opioid drugs include full agonists, partial agonists, and antagonists–measures of intrinsic activity or efficacy. Morphine is a full agonist at the l (mu)–opioid receptor, the major analgesic opioid receptor (Table 31–1). Opioids may also differ in receptor binding affinity. For example, morphine exhibits a greater binding affinity at the μ-opioid receptor than does codeine. Other opioid receptor subtypes include c (delta) and j (kappa) receptors. Simple substitution of an allyl group on the nitrogen of the full agonist morphine plus addition of a single hydroxyl group results in naloxone, a strong μ-receptor antagonist. The structures of some of these compounds are shown later in this chapter. Some opioids, eg, nalbuphine, a mixed agonist-antagonist, are capable of producing an agonist (or partial agonist) effect at one opioid receptor subtype and an antagonist effect at another. The receptor-activating properties and affinities of opioid analgesics can be manipulated by pharmaceutical chemistry; in addition, certain opioid analgesics are modified in the liver, resulting in compounds with greater analgesic action. Chemically, the opioids derived from opium are phenanthrene derivatives and include four or more fused rings, while most of the synthetic opioids are simpler molecules.
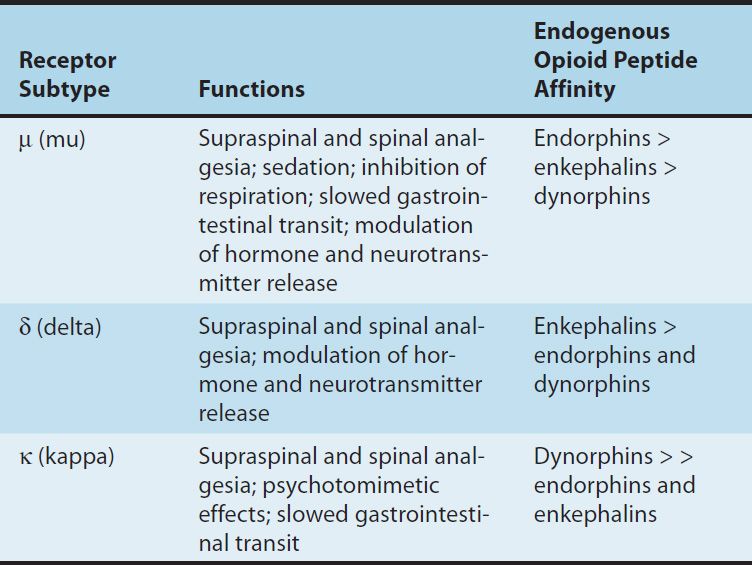
TABLE 31–1 Opioid receptor subtypes, their functions, and their endogenous peptide affinities.
Endogenous Opioid Peptides
Opioid alkaloids (eg, morphine) produce analgesia through actions at central nervous system (CNS) receptors that also respond to certain endogenous peptides with opioid-like pharmacologic properties. The general term currently used for these endogenous substances is endogenous opioid peptides.
Three families of endogenous opioid peptides have been described: the endorphins, the pentapeptide enkephalins (methionine-enkephalin [met-enkephalin] and leucine-enkephalin [leu-enkephalin]), and the dynorphins. These three families of endogenous opioid peptides have overlapping affinities for opioid receptors (Table 31–1).
The endogenous opioid peptides are derived from three precursor proteins: prepro-opiomelanocortin (POMC), preproenkephalin (proenkephalin A), and preprodynorphin (proenkephalin B). POMC contains the met-enkephalin sequence, β-endorphin, and several nonopioid peptides, including adrenocorticotropic hormone (ACTH), β-lipotropin, and melanocyte-stimulating hormone. Preproenkephalin contains six copies of met-enkephalin and one copy of leu-enkephalin. Leu- and met-enkephalin have slightly higher affinity for the δ (delta) than for the μ-opioid receptor (Table 31–1). Preprodynorphin yields several active opioid peptides that contain the leu-enkephalin sequence. These are dynorphin A, dynorphin B, and α and β neoendorphins. Painful stimuli can evoke release of endogenous opioid peptides under the stress associated with pain or the anticipation of pain, and they diminish the perception of pain.
In contrast to the analgesic role of leu- and met-enkephalin, an analgesic action of dynorphin A—through its binding to κ-opioid receptors—remains controversial. Dynorphin A is also found in the dorsal horn of the spinal cord. Increased levels of dynorphin occur in the dorsal horn after tissue injury and inflammation. This elevated dynorphin level is proposed to increase pain and induce a state of long-lasting sensitization and hyperalgesia. The pronociceptive action of dynorphin in the spinal cord appears to be independent of the opioid receptor system. This pronociceptive effect may involve an action via dynorphin A binding to the N-methyl-D-aspartate (NMDA)-receptor complex and possibly to a novel receptor-ligand system homologous to the opioid peptides.
The principal receptor for this novel system is the G protein-coupled orphanin opioid-receptor-like subtype 1 (ORL1). Its endogenous ligand has been termed nociceptin by one group of investigators and orphanin FQ by another group. This ligand-receptor system is currently known as the N/OFQ system. Nociceptin is structurally similar to dynorphin except for the absence of an N-terminal tyrosine; it acts only at the ORL1 receptor, now known as NOP. The N/OFQ system is widely expressed in the CNS and periphery, reflecting its equally diverse biology and pharmacology. As a result of experiments using highly selective NOP receptor ligands, the N/OFQ system has been implicated in both pro- and anti-nociceptive activity as well as in the modulation of drug reward, learning, mood, anxiety, and cough processes, and of parkinsonism.
Pharmacokinetics
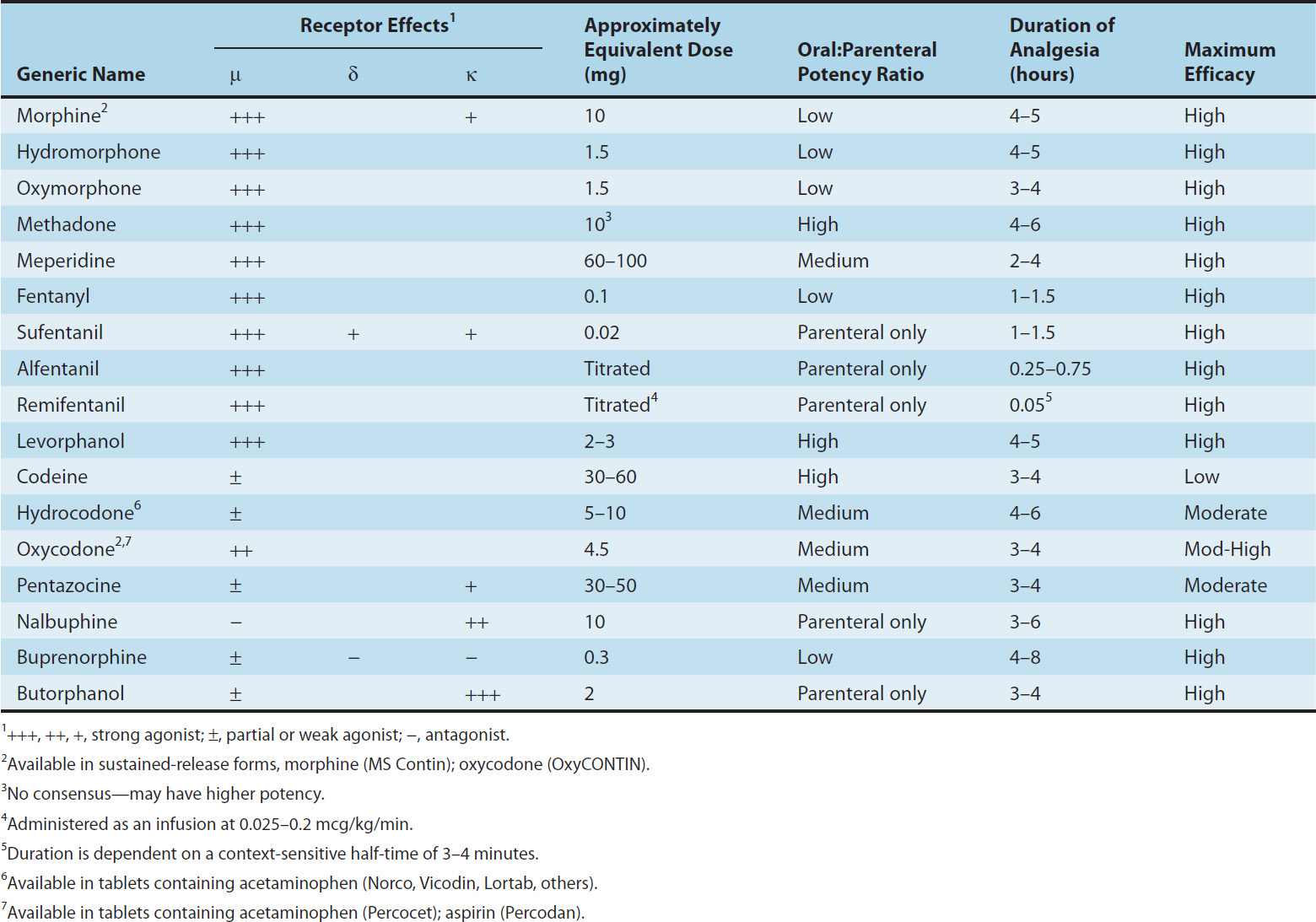
Properties of clinically important opioids are summarized in Table 31–2.
TABLE 31–2 Common opioid analgesics.
A. Absorption
Most opioid analgesics are well absorbed when given by subcutaneous, intramuscular, and oral routes. However, because of the first-pass effect, the oral dose of the opioid (eg, morphine) to elicit a therapeutic effect may need to be much higher than the parenteral dose. As there is considerable interpatient variability in first-pass opioid metabolism, prediction of an effective oral dose is difficult. Certain analgesics such as codeine and oxycodone are effective orally because they have reduced first-pass metabolism. By avoiding first-pass metabolism, nasal insufflation of certain opioids can rapidly result in therapeutic blood levels. Other routes of opioid administration include oral mucosa via lozenges, and the transdermal route via patches. The latter can provide delivery of potent analgesics over days.
B. Distribution
The uptake of opioids by various organs and tissues is a function of both physiologic and chemical factors. Although all opioids bind to plasma proteins with varying affinity, the drugs rapidly leave the blood compartment and localize in highest concentrations in highly perfused tissues such as the brain, lungs, liver, kidneys, and spleen. Drug concentrations in skeletal muscle may be much lower, but this tissue serves as the main reservoir because of its greater bulk. Even though blood flow to fatty tissue is much lower than to the highly perfused tissues, accumulation can be very important, particularly after frequent high-dose administration or continuous infusion of highly lipophilic opioids that are slowly metabolized, eg, fentanyl.
C. Metabolism
The opioids are converted in large part to polar metabolites (mostly glucuronides), which are then readily excreted by the kidneys. For example, morphine, which contains free hydroxyl groups, is primarily conjugated to morphine-3-glucuronide (M3G), a compound with neuroexcitatory properties. The neuroexcitatory effects of M3G do not appear to be mediated by μ receptors and are under further study. In contrast, approximately 10% of morphine is metabolized to morphine- 6-glucuronide (M6G), an active metabolite with analgesic potency four to six times that of its parent compound. However, these relatively polar metabolites have limited ability to cross the blood-brain barrier and probably do not contribute significantly to the usual CNS effects of a single dose of morphine. Importantly, accumulation of these metabolites may produce unexpected adverse effects in patients with renal failure or when exceptionally large doses of morphine are administered or high doses are administered over long periods. This can result in M3G-induced CNS excitation (seizures) or enhanced and prolonged opioid action produced by M6G. CNS uptake of M3G and, to a lesser extent, M6G can be enhanced by co-administration of probenecid or of drugs that inhibit the P-glycoprotein drug transporter.
1. Hepatic P450 metabolism—Hepatic oxidative metabolism is the primary route of degradation of the phenylpiperidine opioids (fentanyl, meperidine, alfentanil, sufentanil) and eventually leaves only small quantities of the parent compound unchanged for excretion. However, accumulation of a demethylated metabolite of meperidine, normeperidine, may occur in patients with decreased renal function and in those receiving multiple high doses of the drug. In high concentrations, normeperidine may cause seizures. In contrast, no active metabolites of fentanyl have been reported. The P450 isozyme CYP3A4 metabolizes fentanyl by N-dealkylation in the liver. CYP3A4 is also present in the mucosa of the small intestine and contributes to the first-pass metabolism of fentanyl when it is taken orally.
Codeine, oxycodone, and hydrocodone undergo metabolism in the liver by P450 isozyme CYP2D6, resulting in the production of metabolites of greater potency. For example, codeine is demethylated to morphine, which is then conjugated. Hydrocodone is metabolized to hydromorphone and, like morphine, hydromorphone is conjugated, yielding hydromorphone-3-glucuronide (H3G), which has CNS excitatory properties. Hydromorphone cannot form a 6-glucuronide metabolite. Similarly, oxycodone is metabolized to oxymorphone, which is then conjugated to oxymorphone-3-glucuronide (O3G).
Genetic polymorphism of CYP2D6 has been documented and linked to the variation in analgesic and adverse responses seen among patients. In contrast, the metabolites of oxycodone and hydrocodone may be of minor consequence; the parent compounds are currently believed to be directly responsible for the majority of their analgesic actions. However, oxycodone and its metabolites can accumulate under conditions of renal failure and have been associated with prolonged action and sedation. In the case of codeine, conversion to morphine may be of greater importance because codeine itself has relatively low affinity for opioid receptors. As a result, some patients (so-called poor metabolizers) may experience no significant analgesic effect. In contrast, there have been case reports of an exaggerated response to codeine due to enhanced metabolic conversion to morphine (ie, ultra rapid metabolizers; see Chapters 4, 5) resulting in respiratory depression and death. For this reason, routine use of codeine, especially in pediatric age groups, is now being eliminated in the United States.
2. Plasma esterase metabolism—Esters (eg, heroin, remifentanil) are rapidly hydrolyzed by common plasma and tissue esterases. Heroin (diacetylmorphine) is hydrolyzed to monoacetylmorphine and finally to morphine, which is then conjugated with glucuronic acid.
D. Excretion
Polar metabolites, including glucuronide conjugates of opioid analgesics, are excreted mainly in the urine. Small amounts of unchanged drug may also be found in the urine. In addition, glucuronide conjugates are found in the bile, but enterohepatic circulation represents only a small portion of the excretory process of these polar metabolites. In patients with renal impairment the effects of active polar metabolites should be considered before the administration of potent opioids such as morphine or hydromorphone—especially when given at high doses—due to the risk of sedation and respiratory depression.
Pharmacodynamics
A. Mechanism of Action
Opioid agonists produce analgesia by binding to specific G protein-coupled receptors that are located in brain and spinal cord regions involved in the transmission and modulation of pain (Figure 31–1). Some effects may be mediated by opioid receptors on peripheral sensory nerve endings.
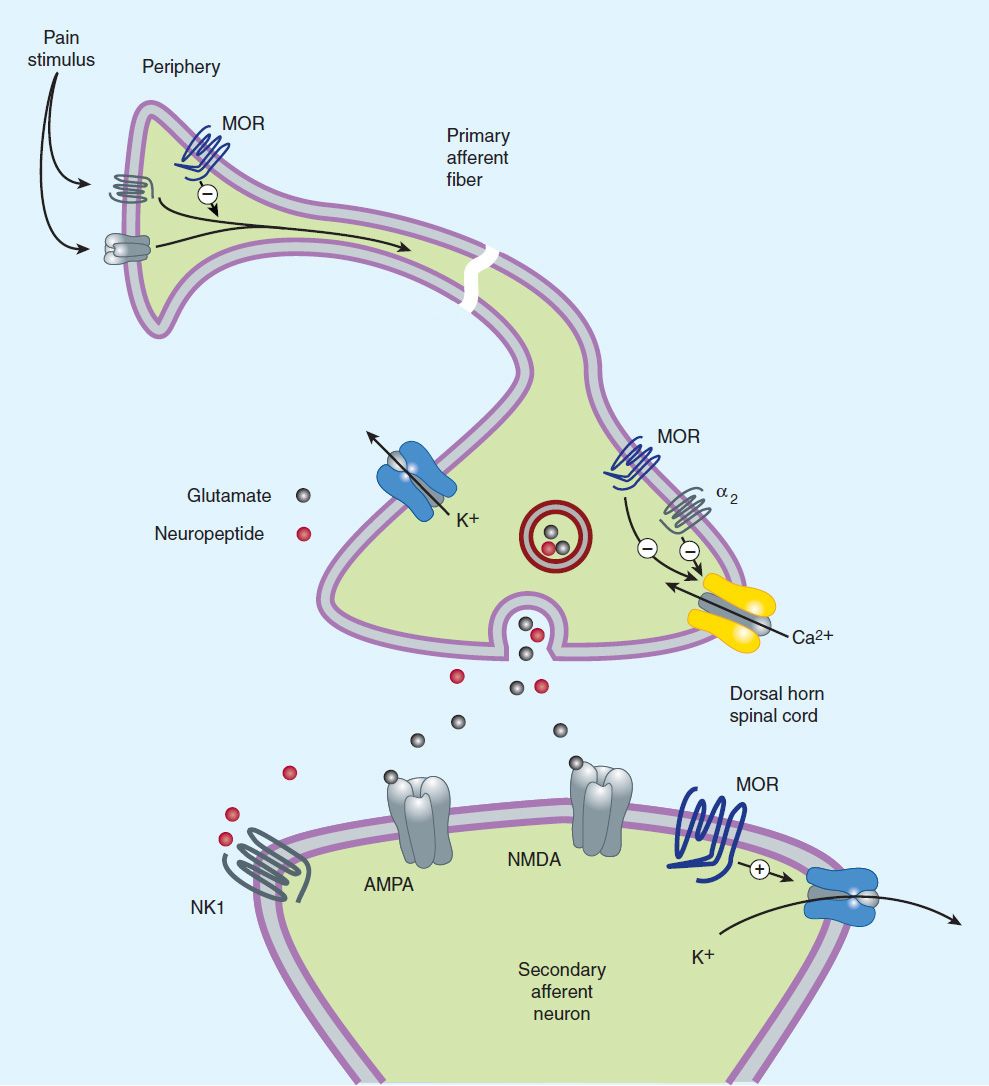
FIGURE 31–1 Potential receptor mechanisms of analgesic drugs. The primary afferent neuron (cell body not shown) originates in the periphery and carries pain signals to the dorsal horn of the spinal cord, where it synapses via glutamate and neuropeptide transmitters with the secondary neuron. Pain stimuli can be attenuated in the periphery (under inflammatory conditions) by opioids acting at μ-opioid receptors (MOR) or blocked in the afferent axon by local anesthetics (not shown). Action potentials reaching the dorsal horn can be attenuated at the presynaptic ending by opioids and by calcium blockers (ziconotide), α2 agonists, and possibly, by drugs that increase synaptic concentrations of norepinephrine by blocking reuptake (tapentadol). Opioids also inhibit the postsynaptic neuron, as do certain neuropeptide antagonists acting at tachykinin (NK1) and other neuropeptide receptors.
1. Receptor types—As noted previously, three major classes of opioid receptors (μ, δ, and κ) have been identified in various nervous system sites and in other tissues (Table 31–1). Each of the three major receptors has now been cloned. All are members of the G protein-coupled family of receptors and show significant amino acid sequence homologies. Multiple receptor subtypes have been proposed based on pharmacologic criteria, including μ1, μ2; δ1, δ2; and κ1, κ2, and κ3. However, genes encoding only one subtype from each of the μ, δ, and κ receptor families have thus far been isolated and characterized. One plausible explanation is that μ-receptor subtypes arise from alternate splice variants of a common gene. This idea has been supported by the identification of receptor splice variants in mice and humans, and a recent report pointed to the selective association of a μ-opioid receptor splice variant (MOR1D) with the induction of itch rather than the suppression of pain.
Since an opioid may function with different potencies as an agonist, partial agonist, or antagonist at more than one receptor class or subtype, it is not surprising that these agents are capable of diverse pharmacologic effects.
2. Cellular actions—At the molecular level, opioid receptors form a family of proteins that physically couple to G proteins and through this interaction affect ion channel gating, modulate intracellular Ca2+ disposition, and alter protein phosphorylation (see Chapter 2). The opioids have two well-established direct Gi/0 protein-coupled actions on neurons: (1) they close voltage-gated Ca2+ channels on presynaptic nerve terminals and thereby reduce transmitter release, and (2) they open K+ channels and hyperpolarize and thus inhibit postsynaptic neurons. Figure 31–1 schematically illustrates these effects. The presynaptic action—depressed transmitter release—has been demonstrated for a large number of neurotransmitters, including glutamate, the principal excitatory amino acid released from nociceptive nerve terminals, as well as acetylcholine, norepinephrine, serotonin, and substance P.
3. Relation of physiologic effects to receptor type—The majority of currently available opioid analgesics act primarily at the μ-opioid receptor (Table 31–2). Analgesia and the euphoriant, respiratory depressant, and physical dependence properties of morphine result principally from actions at μ receptors. In fact, the μ receptor was originally defined using the relative potencies for clinical analgesia of a series of opioid alkaloids. However, opioid analgesic effects are complex and include interaction with δ and κ receptors. This is supported in part by the study of genetic knockouts of the μ, δ, and κ genes in mice. The development of μ-receptor–selective agonists could be clinically useful if their side-effect profiles (respiratory depression, risk of dependence) were more favorable than those found with current μ-receptor agonists, such as morphine. Although morphine does act at κ and δ receptor sites, it is unclear to what extent this contributes to its analgesic action. The endogenous opioid peptides differ from most of the alkaloids in their affinity for the δ and κ receptors (Table 31–1).
In an effort to develop opioid analgesics with a reduced incidence of respiratory depression or propensity for addiction and dependence, compounds that show preference for κ opioid receptors have been developed. Butorphanol and nalbuphine have shown some clinical success as analgesics, but they can cause dysphoric reactions and have limited potency. It is interesting that butorphanol has also been shown to cause significantly greater analgesia in women than in men. In fact, gender-based differences in analgesia mediated by μ- and δ-receptor activation have been widely reported.
4. Receptor distribution and neural mechanisms of analgesia—Opioid receptor binding sites have been localized autoradiographically with high-affinity radioligands and with antibodies to unique peptide sequences in each receptor subtype. All three major receptors are present in high concentrations in the dorsal horn of the spinal cord. Receptors are present both on spinal cord pain transmission neurons and on the primary afferents that relay the pain message to them (Figure 31–2, sites A and B). Although opioid agonists directly inhibit dorsal horn pain transmission neurons, they also inhibit the release of excitatory transmitters from the primary afferents. Although there are reports that heterodimerization of the μ-opioid and δ-opioid receptors contributes to μ-agonist efficacy (eg, inhibition of presynaptic voltage-gated calcium channel activity), a recent study using a transgenic mouse that expresses a δ-receptor–enhanced green fluorescent protein (eGFP) fusion protein showed little overlap of μ receptor and δ receptor in dorsal root ganglion neurons. Importantly, the μ receptor is associated with TRPV1 and peptide (substance P)-expressing nociceptors, whereas δ-receptor expression predominates in the non-peptidergic population of nociceptors, including many primary afferents with myelinated axons. This finding is consistent with the action of intrathecal μ-receptor– and δ-receptor–selective ligands that are found to block heat versus mechanical pain processing, respectively. Very recently, an association of the δ but not the μ receptor with large diameter mechanoreceptive afferents has been described. To what extent the differential expression of the μ receptor and δ receptor in the dorsal root ganglia is characteristic of neurons throughout the CNS remains to be determined.
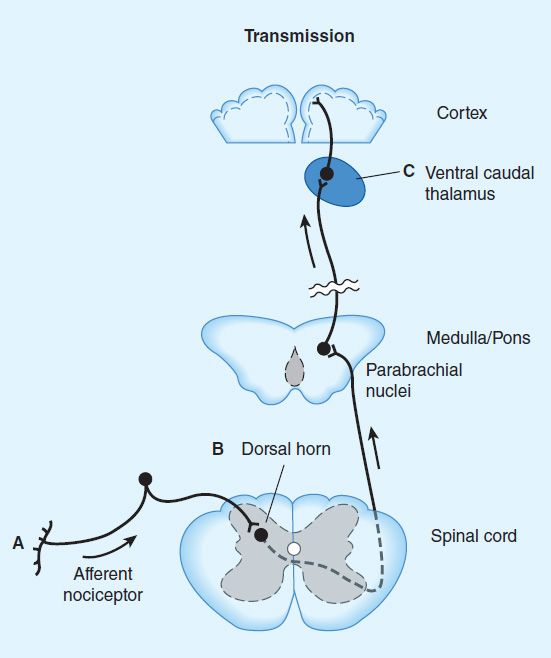
FIGURE 31–2 Putative sites of action of opioid analgesics. Sites of action on the afferent pain transmission pathway from the periphery to the higher centers are shown. A: Direct action of opioids on inflamed or damaged peripheral tissues (see Figure 31–1 for detail). B: Inhibition also occurs in the spinal cord (see Figure 31–1). C: Possible sites of action in the thalamus.
The fact that opioids exert a powerful analgesic effect directly on the spinal cord has been exploited clinically by direct application of opioid agonists to the spinal cord. This spinal action provides a regional analgesic effect while reducing the unwanted respiratory depression, nausea and vomiting, and sedation that may occur from the supraspinal actions of systemically administered opioids.
Under most circumstances, opioids are given systemically and thus act simultaneously at multiple sites. These include not only the ascending pathways of pain transmission beginning with specialized peripheral sensory terminals that transduce painful stimuli (Figure 31–2) but also descending (modulatory) pathways (Figure 31–3). At these sites as at others, opioids directly inhibit neurons; yet this action results in the activation of descending inhibitory neurons that send processes to the spinal cord and inhibit pain transmission neurons. This activation has been shown to result from the inhibition of inhibitory neurons in several locations (Figure 31–4). Taken together, interactions at these sites increase the overall analgesic effect of opioid agonists.
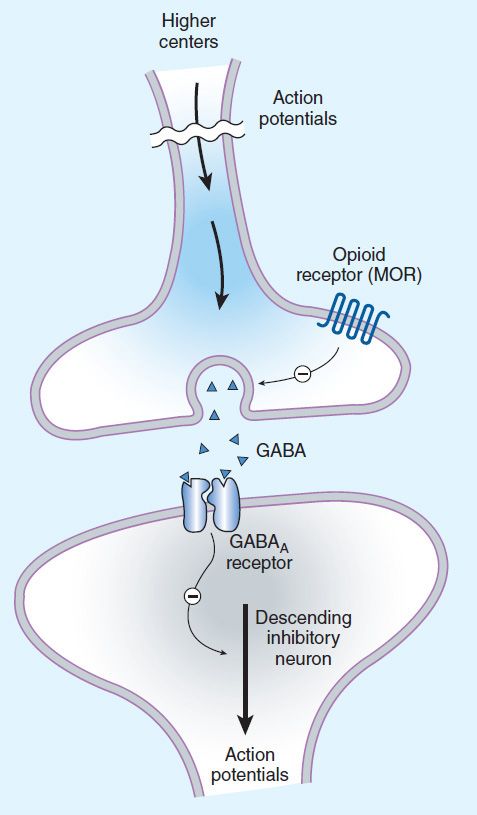
FIGURE 31–3 Brainstem local circuitry underlying the modulating effect of μ-opioid receptor (MOR)–mediated analgesia on descending pathways. The pain-inhibitory neuron is indirectly activated by opioids (exogenous or endogenous), which inhibit an inhibitory (GABAergic) interneuron. This results in enhanced inhibition of nociceptive processing in the dorsal horn of the spinal cord (see Figure 31–4).
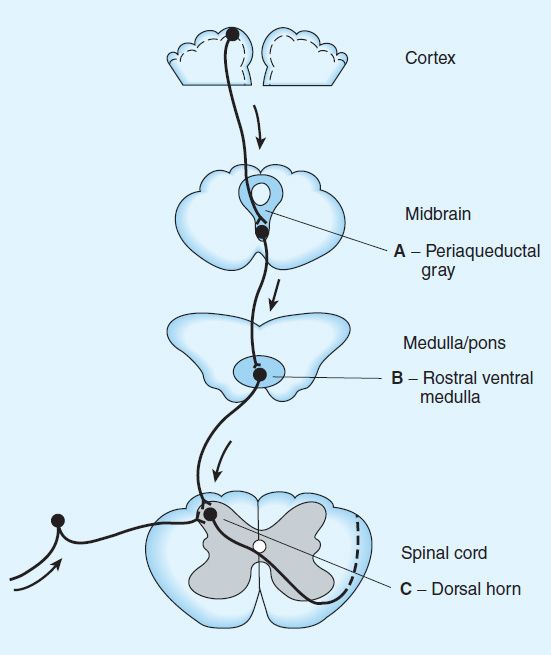
FIGURE 31–4 Opioid analgesic action on the descending inhibitory pathway. Sites of action of opioids on pain-modulating neurons in the midbrain and medulla including the midbrain periaqueductal gray area (A), rostral ventral medulla (B), and the locus caeruleus indirectly control pain transmission pathways by enhancing descending inhibition to the dorsal horn (C).
When pain-relieving opioid drugs are given systemically, they presumably act upon neuronal circuits normally regulated by endogenous opioid peptides and part of the pain-relieving action of exogenous opioids may involve the release of endogenous opioid peptides. For example, an exogenous opioid agonist (eg, morphine) may act primarily and directly at the μ receptor, but this action may evoke the release of endogenous opioids that additionally act at δ and κ receptors. Thus, even a receptor-selective ligand can initiate a complex sequence of events involving multiple synapses, transmitters, and receptor types.
Animal and human clinical studies demonstrate that both endogenous and exogenous opioids can also produce analgesia at sites outside the CNS. Pain associated with inflammation seems especially sensitive to these peripheral opioid actions. The presence of functional μ receptors on the peripheral terminals of sensory neurons supports this hypothesis. Furthermore, activation of peripheral μ receptors results in a decrease in sensory neuron activity and transmitter release. The endogenous release of β-endorphin produced by immune cells within injured or inflamed tissue represents one source of physiologic peripheral μ-receptor activation. Intra-articular administration of opioids, eg, following arthroscopic knee surgery, has shown clinical benefit for up to 24 hours. For this reason opioids selective for a peripheral site of action may be useful adjuncts in the treatment of inflammatory pain (see Box: Ion Channels & Novel Analgesic Targets). Such compounds could have the additional benefit of reducing unwanted effects such as nausea.
5. Tolerance and dependence—With frequently repeated therapeutic doses of morphine or its surrogates, there is a gradual loss in effectiveness; this loss of effectiveness is termed tolerance. To reproduce the original response, a larger dose must be administered. Along with tolerance, physical dependence develops. Physical dependence is defined as a characteristic withdrawal or abstinence syndrome when a drug is stopped or an antagonist is administered (see also Chapter 32).
The mechanism of development of opioid tolerance and physical dependence is poorly understood, but persistent activation of μ receptors such as occurs with the treatment of severe chronic pain appears to play a primary role in its induction and maintenance. Current concepts have shifted away from tolerance being driven by a simple up-regulation of the cyclic adenosine monophosphate (cAMP) system. Although this process is associated with tolerance, it is not sufficient to explain it. A second hypothesis for the development of opioid tolerance and dependence is based on the concept of receptor recycling. Normally, activation of μ receptors by endogenous ligands results in receptor endocytosis followed by resensitization and recycling of the receptor to the plasma membrane (see Chapter 2). However, using genetically modified mice, research now shows that the failure of morphine to induce endocytosis of the μ-opioid receptor is an important component of tolerance and dependence. In further support of this idea, methadone, a μ-receptor agonist used for the treatment of opioid tolerance and dependence, induces receptor endocytosis. This suggests that maintenance of normal sensitivity of μ receptors requires reactivation by endocytosis and recycling.
The concept of receptor uncoupling has also gained prominence. Under this hypothesis, tolerance results from a dysfunction of structural interactions between the μ receptor and G proteins, second-messenger systems, and their target ion channels. Uncoupling and recoupling of μ receptor function is likely linked to receptor recycling. Moreover, the NMDA-receptor ion channel complex has been shown to play a critical role in tolerance development and maintenance. Consistent with this hypothesis, NMDA-receptor antagonists such as ketamine can block tolerance development. Although a role in endocytosis is not yet clearly defined, the development of novel NMDA-receptor antagonists or other strategies to recouple μ receptors to their target ion channels provides hope for achieving a clinically effective means to prevent or reverse opioid analgesic tolerance.
6. Opioid-induced hyperalgesia—In addition to the development of tolerance, persistent administration of opioid analgesics can increase the sensation of pain, resulting in a state of hyperalgesia. This phenomenon can be produced with several opioid analgesics, including morphine, fentanyl, and remifentanil. Spinal dynorphin and activation of the bradykinin and NMDA receptors have emerged as important candidates for the mediation of opioid-induced hyperalgesia. This is one more reason why the use of opioids for chronic pain is controversial.
B. Organ System Effects of Morphine and Its Surrogates
The actions described below for morphine, the prototypic opioid agonist, can also be observed with other opioid agonists, partial agonists, and those with mixed receptor effects. Characteristics of specific members of these groups are discussed below.
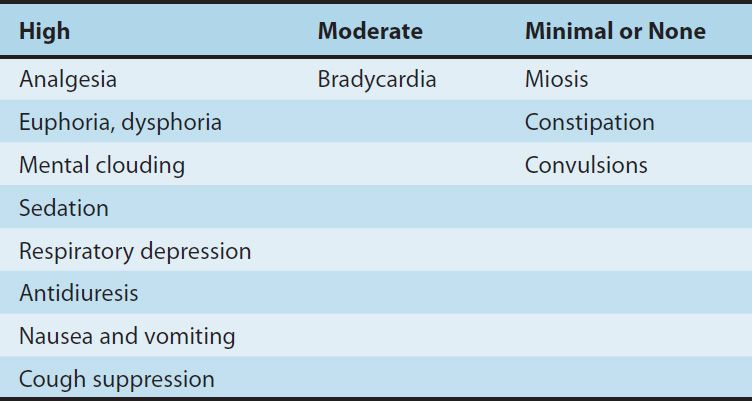
1. Central nervous system effects—The principal effects of opioid analgesics with affinity for μ receptors are on the CNS; the more important ones include analgesia, euphoria, sedation, and respiratory depression. With repeated use, a high degree of tolerance occurs to all of these effects (Table 31–3).
TABLE 31–3 Degrees of tolerance that may develop to some of the effects of the opioids.
Even the most severe acute pain (lasting hours to days) can usually be well controlled—with significant but tolerable adverse effects—using currently available analgesics, especially the opioids. Chronic pain (lasting weeks to months), however, is not very satisfactorily managed with opioids. It is now known that in chronic pain, receptors on sensory nerve terminals in the periphery contribute to increased excitability of sensory nerve endings (peripheral sensitization). The hyperexcitable sensory neuron bombards the spinal cord, leading to increased excitability and synaptic alterations in the dorsal horn (central sensitization). Such changes are likely important contributors to chronic inflammatory and neuropathic pain states.
In the effort to discover better analgesic drugs for chronic pain, renewed attention is being paid to the molecular basis of peripheral sensory transduction. Potentially important ion channels associated with the primary afferent nociceptor include members of the transient receptor potential family, notably the capsaicin receptor, TRPV1 (which is activated by multiple noxious stimuli such as heat, protons, and products of inflammation) as well as TRPA1, activated by inflammatory mediators; and P2X receptors (which are responsive to purines released from tissue damage). Special subtypes of voltage-gated sodium channels (Nav 1.7, 1.8, 1.9) are uniquely associated with nociceptive neurons in dorsal root ganglia. Lidocaine and mexiletine, which are useful in some chronic pain states, may act by blocking this class of channels. Certain centipede toxins appear to selectively inhibit Nav 1.7 channels and may also be useful in the treatment of chronic pain. Genetic polymorphisms of Nav 1.7 are associated with either absence or predisposition to pain. Because of the importance of their peripheral sites of action, therapeutic strategies that deliver agents that block peripheral pain transduction or transmission have been introduced in the form of transdermal patches and balms. In addition, products that systemically target peripheral TRPV1, TRPA1 and sodium channel function are in development.
Ziconotide, a blocker of voltage-gated N-type calcium channels, is approved for intrathecal analgesia in patients with refractory chronic pain. Ziconotide is a synthetic peptide related to the marine snail toxin ω-conotoxin, which selectively blocks N-type calcium channels. Gabapentin/pregabalin, anticonvulsant analogs of GABA (see Chapter 24) that are effective treatments for neuropathic (nerve injury) pain act at the α2δ1 subunit of voltage-gated calcium channels. N-methyl-d-aspartate (NMDA) receptors appear to play a very important role in central sensitization at both spinal and supraspinal levels. Although certain NMDA antagonists have demonstrated analgesic activity (eg, ketamine), it has been difficult to find agents with an acceptably low profile of adverse effects or neurotoxicity. However, ketamine infused at very small doses improves analgesia and can reduce opioid requirements under conditions of opioid tolerance, eg, after major abdominal and spinal surgery. GABA and acetylcholine (through nicotinic receptors) appear to control the central synaptic release of several transmitters involved in nociception. Nicotine itself and certain nicotine analogs cause analgesia, and their use for postoperative analgesia is under investigation. Use of antibodies that bind nerve growth factor (NGF) has been shown to block inflammatory and back pain and is awaiting FDA approval. Finally, work on cannabinoids and vanilloids and their receptors suggest that Δ9- tetrahydrocannabinol, which acts primarily on CB1 cannabinoid receptors, can synergize with μ-receptor analgesics and interact with the TRPV1 capsaicin receptor to produce analgesia under certain circumstances.
As our understanding of peripheral and central pain transduction improves, additional therapeutic targets and strategies will become available. Combined with our present knowledge of opioid analgesics, a “multimodal” approach to pain therapy is emerging. Multimodal analgesia involves the administration of multiple agents (eg, NSAIDs, gabapentinoids, selective norepinephrine receptor inhibitors, etc) with complementary mechanisms of action to provide analgesia that is superior to that provided by an individual compound. Another benefit of multimodal analgesia is reduced opioid requirements with fewer adverse effects.
a. Analgesia—
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


