Type
Cytogenetic change(s)
Genes affected
Pathway type
CML
t(9;22)
BCR–ABL1
TK growth
+8, del17p (CE)
PV, PMF, ET
XY/XX
JAK2
Cytokine receptors
+9, +1/1q (CE)
CEL/MPN-U
XY/XX
FIP1L1–PDGFRA
RTK
t(8p11;var)
FGFR1-various
CMML, MPN-U
t(5;12)(q31-q33;p12)
ETV6–PDGFRB
t(5q33;var)
PDGFRB-various
Table 44.2
Categories of myelodysplastic syndrome and their molecular alterations
Type | Definition |
|---|---|
Refractory cytopenia with unilineage dysplasia, including refractory anemia, neutropenia, and thrombocytopenia | Cytopenia and marrow morphologic dysplasia limited to one of the three primary lineages |
Blasts: <5 % BM, <1 % blood | |
Refractory Anemia with ringed sideroblasts (RS) | Anemia, ≥15 % BM erythroid precursors meeting definition of RS |
Blasts: <5 % BM, <1 % blood | |
Myelodysplastic syndrome with isolated del(5q) | Anemia, high platelet count and isolated deletion(s) of chromosome 5q |
Blasts: <5 % BM, <1 % blood | |
Refractory cytopenias with multilineage dysplasia +/− RS | Cytopenias, morphologic dysplasia identified in two or more lineages |
Blasts: <5 % BM, <1 % blood | |
Refractory anemia with excess blasts-I | Cytopenias |
Blasts: 5–9 % BM and <5 % blood | |
Refractory anemia with excess blasts-II | Cytopenias |
Blasts: 10–19 % BM and/or >5 % blood, Auer rods can be present | |
Myelodysplasia, unclassifiable | Cytopenias |
Blasts: <5 % BM, <1 % blood | |
Often used for those cases showing some features of myeloproliferative neoplasms, e.g., increased basophils, eosinophils, and/or fibrosis | |
Chronic myelomonocytic leukemia | 1 × 109/L monocytes in blood, with marrow dysplasia |
Mixed disorder with more of a myeloproliferative pattern in some cases |
Besides affecting the myeloid compartment of the hematopoietic system, MPN and MDS share many overlapping morphologic, genetic, and immunophenotypic features (Table 44.3), with the distinction based on the dominant presenting feature. MPNs have increased numbers of blood components due to abnormal proliferation. In contrast, MDS typically presents with decreased numbers of blood components due to ineffective hematopoiesis. An overlap category of myelodysplastic/myeloproliferative neoplasms is recognized when there are features of both entities. Molecular testing in MPNs has become established as the standard of care for diagnosis, monitoring for residual disease, establishing prognosis, and therapeutic decision making. In MDS, the role of molecular testing is currently more limited with cytogenetic studies representing the primary modality used for subclassification and prognostic stratification. However, mutational and epigenetic studies are poised to become part of the routine workup of MDS in the near future.
Table 44.3
Shared features between MDS and MPNs
Immunophenotypic similarities | Loss/decreased expression of pan-myeloid antigens |
Aberrant upregulation of adhesion molecules (e.g., CD56) | |
Cytogenetic similarities | Acquisition of +8 with progression |
Acquisition of TP53 mutations with progression | |
LOH/UPD involving myeloid regulatory loci | |
Molecular similarities | Mutations in the epigenetic regulators including IDH1, IDH2, TET2, and DNMT3A |
Mutations in RAS genes and myeloid transcriptional regulators such as ASXL1 |
Indications for Testing
Myeloproliferative Disorders
Elevated white blood cells (leukocytosis), red blood cells (erythrocytosis), or platelets (thrombocytosis), and/or splenomegaly are the hallmarks of MPNs. However, these changes also can represent reactive phenomena, as a manifestation of the normal bone marrow (BM) stress response to infection, trauma, or other types of injury. Because distinction of reactive and neoplastic expansions can be difficult on clinical and morphologic features, molecular and cytogenetic testing can lead to a definitive diagnosis (Table 44.1). The most common MPNs are CML, essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). These entities can have overlapping morphologic and clinical characteristics [2]. A variety of less common MPNs have been recognized in recent years and can be definitively diagnosed by the presence of characteristic cytogenetic or molecular changes.
Chronic Myelogenous Leukemia
Patients with CML typically present with neutrophilia and variable basophilia and eosinophilia. Although some patients are asymptomatic and diagnosed following routine blood work, presentations with fatigue and/or splenomegaly are common. Left untreated, many patients with CML eventually progress to an accelerated phase and then to blast crisis which can be indistinguishable from acute myeloid leukemia (AML).
The medical advances made in the diagnosis and treatment of CML represent some of the greatest achievements in molecular medicine to date. CML was the first malignancy noted to have a defining genetic abnormality when the Philadelphia chromosome (Ph), a translocation between chromosomes 9 and 22, t(9;22) (q34;q11), was identified [1]. Subsequently, the Ph chromosome was identified as the juxtaposition of the breakpoint cluster region (BCR) gene on chromosome 22 with the Abelson tyrosine kinase (ABL1) gene on chromosome 9 (Fig. 44.1a), resulting in relocalization of an altered ABL1 protein from the nucleus to the cytoplasm, which produces constitutive activation of the ABL growth signaling pathway. The BCR–ABL1 gene fusion is found in all cases of CML and in about 20–30 % of adult and 2–10 % of childhood B lymphoblastic leukemia (B-ALL) [3].
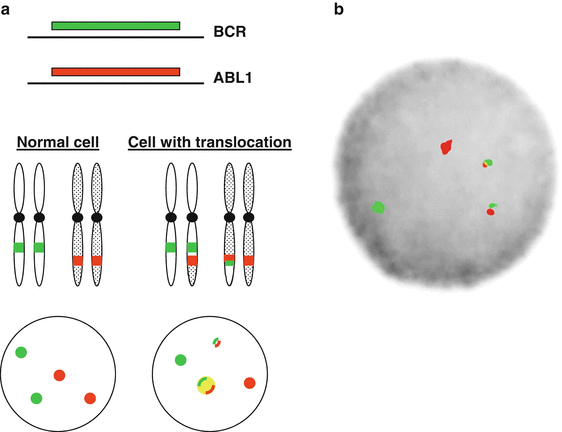
Figure 44.1
BCR–ABL1 gene rearrangement detected by fluorescence in situ hybridization (FISH). (a) Schematic of fusion FISH with orange-labeled probes for ABL1 gene on chromosome 9 and green-labeled probes on the BCR gene on chromosome 22. (b) Image from a CML cell showing the t(9;22)
The differential diagnosis of CML includes benign leukocytosis/leukemoid reaction and the rare chronic neutrophilic leukemia (CNL) and its related atypical chronic myeloid leukemia (aCML). Although CNL and aCML typically lack the basophilia associated with CML other features may be similar. In 2013, cases of CNL/aCML were shown to have a high frequency of activating and truncation mutations in the colony-stimulating factor 3 receptor gene (CSF3R) [4].
Essential Thrombocythemia, Primary Myelofibrosis, and Polycythemia Vera
Based on clinical features, ET, PMF, and PV are closely related MPNs, distinguished primarily by their dominant presenting features and clinical course. This close clinical relationship was explained in 2005 when several groups independently identified a point mutation in the JAK2 tyrosine kinase gene as a recurrent abnormality in these three MPNs, and some cases of chronic myelomonocytic leukemia (CMML). The activating JAK2 mutation, V617F, is detected in up to 95 % of cases of PV, and approximately 50 % of ET and PMF cases (Table 44.4). A subset of PV, but not PMF or ET, has activating point mutations or in-frame duplications in exon 12 of JAK2. Among PMF and ET without V617F, activating mutations in the thrombopoietin receptor gene (MPL) are seen in approximately 10 % of cases. Because the JAK2 kinase links multiple cytokine receptors, including the erythropoietin receptor, the thrombopoietin receptor (MPL) and the granulocyte-monocyte cytokine colony stimulating factor receptor (CD116), these mutations define this group of MPNs as having JAK-linked dysregulated cytokine growth factor receptor pathways.
Table 44.4
Frequency of mutations in different MPNs
Mutation type | PV | ET | PMF | CMML |
|---|---|---|---|---|
JAK2 V617F | 95 % | 50 % | 40 % | 5 % |
JAK2 exon 12/13 | 5 % | ND | ND | <1 % |
MPL codon 505 or 515 | ND | 5 % | 5 % | ND |
KRAS–NRAS codons 12/13/61 | ND | ND | <1 | 30–40 % |
Differences in the features and behavior of JAK2–MPL-mutated MPNs are related to the spectrum of other coexisting somatic mutations in any particular case [5], to genetic polymorphisms in the cytokine genes of the affected individual [6], and to the varying gene dosage and level of expression of the mutated JAK2 or MPL. As a result of loss of heterozygosity or mutation of both alleles, JAK2 V617F can be present in up to several copies per cell, and cases with a higher V617F allele burden often manifest as PV or PMF rather than as ET. In contrast to patients with the V617F mutation, PV patients with JAK2 exon 12 mutations tend to present at a younger age, and with higher hematocrit, lower platelet count, and lower white blood cell (WBC) count. ET and PMF patients with MPL mutations tend to show different patterns of clonal evolution than JAK2-mutated cases, with the common karyotypic findings of gains of chromosome 1 in ET with MPL mutations and chromosome 9 in PMF with MPL mutations [7]. Germline JAK2 and MPL mutations continue to be discovered in families with predisposition to the development of MPNs [8].
Calreticulin gene (CALR) mutations occur in 50–71 % of ET cases, and 56–88 % of PMF cases that are negative for JAK2 and MPL mutations [9–12]. These mutations include a variety of insertion and deletions located in exon 9 of CALR that shift the translational reading frame and produce a common novel C-terminal peptide sequence. Mutated CALR may differentially influence signaling through the JAK-STAT pathway. CALR mutations are largely mutually exclusive with JAK2 and MPL mutations and are not found in PV, CML, AML, or most cases of MDS. CALR-mutated PMF is prognostically distinct from MPL– and JAK2-mutated MPNs and from those cases that lack all three mutations [9–14]. Therefore, testing for CALR mutations can assist in the diagnosis of MPN, and provide a genetic marker for monitoring response to therapy.
Myeloproliferative Neoplasms Associated with Eosinophilia
Most causes of significant eosinophilia are reactive or secondary to chronic parasitic infections or due to cytokine-producing malignancies, particularly T-cell lymphoma, Hodgkin lymphoma, and some carcinomas. However, a group of rare MPNs present with eosinophilia and immature/dysplastic myelopoiesis and are characterized by gene fusions involving a variety of growth factor genes (Table 44.1). The majority show fusion of the FGFR1 gene to a variety of other genes, thereby producing ligand-independent FGFR1 activation. Another type has a chromosome 4 interstitial cryptic FIP1L1–PDGFRA fusion that activates the PDFRA kinase, with the neoplasms showing dramatic response to imatinib treatment, similar to CML. A very rare PDGFRB-translocation syndrome produces a similar MPN due to fusion with ETV6 or a variety of other partner genes.
Myelodysplastic/Myeloproliferative Neoplasms
The most common of the mixed myeloid disorders is CMML, which can present with a high WBC count resembling acute monocytic leukemia, or with a low WBC count associated with dyspoietic BM findings. This variability in presentation underlies a heterogeneous molecular pathogenesis. RAS gene mutations are identified in the vast majority of cases with hyperproliferative presentations, and a variety of cytogenetic and somatic mutations, including TET2 and JAK2 V617F, are seen in other cases [15].
Myelodysplastic Syndromes
Decreased numbers of WBCs (leukopenia), platelets (thrombocytopenia), and erythrocytes (anemia) are among the most common abnormal laboratory findings in patients with MDS. In younger patients, these cytopenias often have a nonneoplastic etiology such as immune-mediated marrow suppression, autoimmune disease, acute bacterial and viral infections, nutritional deficiency (e.g., B12, folate or iron), or toxin exposure. However, with advancing age, cytopenias are increasingly related to genetically mediated BM failure or MDS. The dysplastic changes of MDS can be superimposed upon other causes of marrow suppression, exacerbating the ineffective hematopoiesis. In these cases, abnormal blood cells show impaired maturation and are destroyed by apoptosis before being released into the bloodstream. These genetically abnormal marrow components often have an atypical morphology (dysplasia) that can be recognized in a BM aspirate smear.
Available Assays and Laboratory Issues
Chronic Myelogenous Leukemia
BCR–ABL1 Detection
In CML, most of the BCR–ABL1 translocations involve the major breakpoint cluster region (M-bcr) adjacent to exons 12–16 of BCR (formerly called exons b1–b5) and result in either the e13a2 or e14a2 BCR–ABL1 gene fusions (or both transcripts), which encode closely related 210 kD fusion proteins (p210). In contrast, in Ph chromosome-positive (Ph+) B-ALL, 50–65 % of the BCR breakpoints arise at the minor breakpoint cluster region (m-bcr), adjacent to exons 1 and 2 of BCR, with the BCR–ABL1 transcript containing the e1a2 junction, which is translated into the p190 BCR-ABL1 fusion protein. Rarely, usually in CML with monocytosis and neutrophilia, the breakpoint occurs in the μ-BCR region (exons 17–20), resulting in a larger p230 fusion BCR-ABL1 protein.
The BCR–ABL1 fusion gene can be detected by several methods. The Ph chromosome can be visualized by conventional G-banded karyotyping with a false-negative rate of < 10 %, which is usually related to more complex fusions that obscure the breakpoints involved. However, karyotyping has a low sensitivity that is not acceptable for minimal residual disease (MRD) monitoring given that only 20 metaphases are usually analyzed (i.e., a maximal sensitivity of 5 %).
Fluorescence in situ hybridization (FISH) on interphase cells is another method to detect the BCR–ABL1 fusion (Fig. 44.1). Dual-color, dual-fusion probes are typically used and can identify variant breakpoints or three-way translocations that do not result in recognizable Ph+ metaphases. Given an adequate sample, the false-negative rate for FISH is much less than 1 %. The use of FISH for MRD monitoring after completion of therapy is more limited given that the maximal sensitivity is approximately 1 % Ph+ cells, given that only several hundred cells are typically counted [16].
Other molecular methods can be used for detecting BCR–ABL1 fusions, including PCR from genomic DNA, Southern blotting, or northern blotting. However, the vast majority of clinical molecular laboratories detect the BCR–ABL1 fusion gene by reverse transcription polymerase chain reaction (RT-PCR). Qualitative RT-PCR has been largely replaced by quantitative/real-time PCR (RT-qPCR) methods using an allele-specific PCR approach with primers directed separately against the M-bcr and m-bcr regions (p210 and p190 isoforms, respectively).
BCR–ABL1 RT-PCR Assay Standardization and the International Scale
Essentially all RT-qPCR BCR–ABL1 assays normalize the amount of fusion transcript to the level of a control transcript to allow comparison across different levels of input positive cells. This approach also permits assessment of RNA quality, because low levels of the control gene likely indicate a degraded sample. Commonly used genes for normalization include GAPDH, ACTB, BCR, GUSB, and ABL1 [17]. These control genes are expressed at very different levels resulting in highly variable BCR–ABL1 fusion to control transcript values. This variability between assays using different control transcripts makes comparison of results from different laboratories difficult. Additionally, laboratories use a variety of RNA extraction methods, different reverse transcription protocols, and have different reporting criteria.
Standardization of RT-qPCR assays has become a high priority because molecular milestones of BCR–ABL1 transcript levels are now the basis of treatment decision-making for CML. The first advance in standardization, led by the Europe Against Cancer (EAC) initiative, was the development of well-validated RT-qPCR primers and probes, as well as standardized PCR conditions for BCR–ABL1 assays [18]. The second effort, beginning with the European LeukemiaNet collaborative, developed consensus on measurement of therapeutic milestones in CML (Table 44.5) [19]. This effort reached fruition in 2012 and established the International Scale (IS) whereby individual laboratories could adjust their internal BCR–ABL1/control transcript ratios to the molecular milestones based on standard calibrators [20, 21].
Table 44.5
Definitions and therapeutic milestones for BCR–ABL1 disease response
Definitions | ||
Complete hematologic response (CHR) | WBC count < 10 × 109/LNormal WBC differential≤1% circulating immature cellsPlatelet count < 450 × 109/LNo signs and symptoms of CML | |
Major molecular response (MMR) | >3-log reduction in BCR–ABL1 transcript levels (0.1 % IS) | |
Complete molecular response (CMR) | PCR negative | |
Complete cytogenetic response (CCyR) | No Ph detected | |
Partial/major cytogenetic response (PCyR) | 1–35 % Ph+ cells | |
Therapeutic milestones | Suboptimal | Failure |
3 months on therapy | Less than CHR | No HR |
6 months | Less than PCyR | Less than CHR |
12 months | Less than CCyR | Less than PCyR |
Not approaching MMR | ||
18 months | Less than MMR | Less than CCyR |
Therapy and Treatment Response Monitoring in CML
With an understanding of the molecular basis of CML, the design and synthesis of small molecules that could inhibit the kinase activity of ABL1 was attempted. This resulted in US Food and Drug Administration (FDA) approval of the kinase inhibitor imatinib mesylate (also called ST1571) in 1999. Before the discovery of imatinib, the widely used therapeutic options for CML were interferon-α with or without cytarabine, hydroxyurea, or allogeneic stem cell transplant (ASCT). With results first published in 2003, the pivotal International Randomized Interferon vs ST1571 (IRIS) study established imatinib as the frontline therapy for newly diagnosed CML, with superior responses and compliance rates compared to interferon/cytarabine [22, 23]. Subsequently, second generation tyrosine kinase inhibitors (TKI) that successfully target BCR-ABL1 were introduced, particularly nilotinib and dasatinib that were both approved as frontline therapy by the FDA in 2010.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


