Figure 25.1
Schematic representation of the MEN1 gene indicating the intron–exon organization, as well as the relative location, type, and distribution of germline mutations characterized in patients with MEN1. Functional domains of menin that interact with JUND, SMAD3, and NFKB are indicated below the gene map. Regions 1 and 2 within exon 10 note the locations of the nuclear localization signals 1 and 2 of the menin protein. From Wautot V, Vercherat C, Lespinasse J, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasias type 1: search for correlations between phenotype and the functional domains of the MEN1 protein. Hum Mutat 2002;20:35–47, copyright © 2002 [3]. Reprinted with permission from John Wiley and Sons
Clinical Utility of Testing
Genetic testing for MEN1 gene mutations is performed for three main reasons: (1) confirmation of a clinical diagnosis of MEN1, (2) identification of a presymptomatic at-risk relative, and (3) cessation of clinical screening in relatives who do not carry a mutation previously identified in affected family members. Approximately 50 % of patients with MEN1 will die as a result of their disease; thus, genetic screening to identify family members with germline MEN1 mutations may lead to closer monitoring, earlier identification and treatment of tumors, improved outcomes, and longer survival. Confirmation of the diagnosis relies on identification of an MEN1 germline mutation. Primary hyperparathyroidism is present in 100 % of patients with MEN1 by the age of 50 years [14], but parathyroid carcinomas are rarely seen. In addition, pancreatic islet cell adenomas may be observed in about 60 % of patients with MEN1 and are most commonly classified as gastrinomas or insulinomas. Pituitary adenomas also occur in approximately 30 % of MEN1 patients, but pituitary carcinomas are not usually detected. Variable expressivity and reduced penetrance also have been reported in many MEN1 families, making distinction of a familial condition difficult to discern from sporadic tumor occurrence. Absence of a detectable germline MEN1 mutation does not exclude the diagnosis of MEN1. Genetic variability in other genes with which the menin protein interacts also may contribute to variable phenotypes of family members with the same MEN1 allele.
Identification of carriers of MEN1 gene mutations has not been very successful in either predicting or preventing the course of MEN1. The utility of MEN1 mutation detection in directing individual patient care is limited by the absence of very sensitive imaging methods for early detection and treatment of MEN1-associated tumors, coupled with the lack of adequate treatment options for the types of tumors observed in MEN1 [12]. Knowledge of the carrier status is not an indicator for prophylactic surgery as in MEN2; thus, mutation screening in MEN1 patients does not avoid or cure the malignancy but may assist in clinical monitoring and lifestyle decisions. Predictive testing of at-risk relatives or prenatal screening requires that a disease-causing MEN1 mutation has been previously detected in the family. When a family-specific mutation can be identified, the clinical significance of a subsequent negative genetic analysis in an at-risk relative can aid the genetic counselor in reassuring that family member and reducing screening to that of the general population. Targeting therapy to the menin protein has not been attempted because its function and the protein domains required for tumorigenesis have not been identified [13]. However, some small-molecule drug therapies are in development (see Conclusions and Future Directions below).
Available Assays
The mutations in the 9.8 kb genomic DNA of the MEN1 gene that are associated with the MEN1 syndrome are diverse in both type and distribution. The initial molecular pathology screening of affected members of MEN1 families should include analysis of the entire gene. Up to 95 % of patients will have a MEN1 mutation identified [15] when a combination of complete gene sequencing and deletion/duplication analyses is employed. Most approaches incorporate polymerase chain reaction (PCR) amplification of each exon with a survey for sequence variants by dideoxy fingerprinting [4], heteroduplex analysis (HA), single-strand conformation polymorphism (SSCP), or direct DNA sequencing [3, 12, 16]. PCR products from exons showing sequence variants by conformational changes usually are sequenced to identify the specific mutation. These strategies will identify mutations in 80–90 % of MEN1 families [17]. As in most PCR-based screening procedures, some types of mutations can be missed, including large deletions or insertions, point mutations in the 5′ regulatory or untranslated regions, nucleotide changes in the introns, or at the sites of PCR primer annealing, which may adversely affect PCR amplification efficiency. The majority of mutations are nonsense or frameshift mutations that predict expression of a truncated menin protein with loss of function [15]. Missense mutations in MEN1 are primarily clustered in the domains that interact with the transcription factors, JUND (MEN1 codons 1–40, 139–242, 323–428), SMAD3 (distal to MEN1 codon 478), and NFKB (MEN1 codons 276–479), of key cell growth pathways (Fig. 25.1) [18]. Additional analysis using Southern hybridization [19, 20], long-range PCR, quantitative PCR, multiplex ligation-dependent probe amplification, or targeted genomic hybridization arrays will detect larger deletion and duplication mutations in MEN1 families following negative findings of direct DNA sequencing approaches. Clinical laboratories also may use next-generation sequencing methods to detect a broader range of mutations in a single test, including single-nucleotide variants, as well as small and large insertions and deletions. Thirty-seven Clinical Laboratory Improvement Amendments (CLIA)-certified clinical laboratories are listed in the Genetic Testing Registry database (www.ncbi.nlm.nih.gov/gtr/ accessed 6/25/15) as offering clinical testing for MEN1 [21]; while all of these laboratories indicate that DNA sequencing is performed, 25 also indicate that deletion and duplication analysis is included. Linkage analysis methods may be used to define familial haplotypes that confer genetic predisposition to MEN1.
Interpretation of Results
For any clinical molecular genetic test, the ability to identify a specific gene mutation to correlate with the disease status of an affected individual depends on the sensitivity of the assay. A well-defined clinical phenotype is essential for accurate interpretation and reporting of molecular test results to the healthcare professional. Genotype–phenotype correlations are either imperfect or nonexistent for MEN1.
Approximately 95 % of patients with MEN1 will have a germline mutation identified in the MEN1 gene coding for the menin protein [15, 17]. MEN1 germline mutations also have been identified in nearly two-thirds of patients with sporadic MEN1 [12]. There is presently no specific genotype–phenotype association of MEN1 mutations to predict the clinical course or onset of this disease [15, 17]. More than 1,000 different mutations have been described [7], and some have been reported in multiple, independent kindreds. Characterization of these same genetic variations in newly diagnosed individuals supports the role of the mutant allele in the disease etiology and is very helpful for interpretation of the identified variant. As in many genetic syndromes, molecular identification of a pathogenic MEN1 mutation in a family member with clinical features of MEN1 is most significant in the risk assessment of other first-degree relatives. Approximately 20 % of patients with familial isolated hyperparathyroidism, characterized by parathyroid adenoma or hyperplasia in the absence of other endocrine neoplasias, carry germline MEN1 gene mutations [17, 22].
As reviewed by Guo and Sawicki [12], MEN1 mutations are not limited to patients with MEN1. A significant number of sporadic endocrine tumors also may harbor MEN1 mutations but not with the same prevalence as the same types of tumors in the syndromic cases. MEN1 mutations do not provide clinically useful information for sporadic tumor staging [13]. While one third of MEN1 patients will develop pituitary tumors and have germline MEN1 mutations, only about 1 % of sporadic pituitary tumors have MEN1 mutations. The frequency of MEN1 mutations also differs between different types of pancreatic endocrine tumors. In sporadic gastrinomas, the rate of MEN1 mutations is 37 % in contrast to 15 % of sporadic nongastrinoma pancreatic tumors. Insulinomas, another pancreatic endocrine tumor of MEN1, develop in MEN1 knockout mice [23], but MEN1 mutations have not been found in sporadic insulinomas [24]. Clearly the role of menin in familial and sporadic tumors is not the same. Sporadic parathyroid and pituitary adenomas with no detectable MEN1 mutation may be characterized as phenocopies of MEN1, particularly when there is no family history of MEN1 [12].
Information about the penetrance and expressivity of the mutations in MEN1 described to date suggests that carriers of a familial MEN1 mutation have a 100 % risk of developing MEN1 by the age of 60 years [3]. Mutations leading to expression of a truncated menin protein create a loss of functional menin protein. Missense mutations may disrupt menin interaction with cell growth regulatory molecules or its cellular localization. These mutations also may decrease the stability of menin protein and lead to its degradation.
Laboratory Issues
Complete analysis of the MEN1 gene sequence by PCR amplification of each exon and untranslated regions followed by direct DNA sequencing fails to identify mutations in 10–20 % of patients with MEN1. Some MEN1 mutations have been identified repeatedly in apparently unrelated families, suggesting some tendency for mutational hot spots within MEN1, with mutation clusters at codons 83–84, 516, and 210–211 accounting for approximately 12 % of all mutations [7]. Caution must be used in the interpretation of the pathogenic functional consequences of DNA sequence variants. Predicting abnormal protein function from a DNA sequence change is imperfect. Approximately 24 polymorphisms of MEN1 have been reported. For example, Arg171Gln and Ala541Thr produce amino acid substitutions featuring different polar side chains but are not associated with a disease phenotype [12]. Missense mutations in amino acids surrounding these same codons are consistent with MEN1 incidence in affected families [3]. Classification of polymorphisms as benign may change with long-term follow-up or ascertainment of additional MEN1 kindreds. Some frameshift or nonsense mutations do not completely abolish normal protein function. The existence of phenocopies (clinical presentations that may mimic familial disorders) also should be a consideration due to the relatively high frequency of sporadic parathyroid and pituitary adenomas [12], particularly in the absence of a family history of MEN1. Genotype–phenotype prediction may be further complicated by variable expression among family members or different families with the same sequence change or by slow growth of an MEN1 tumor even after a gene mutation has been characterized. Once a sequence change has been documented as present in individuals with clinical features of MEN1 and absent in unaffected individuals, one can be reasonably certain of the significance of the finding as a true disease-associated mutation.
Approximately 70 % of reported mutations are nonsense, frameshift, or splice-site mutations that predict the expression of a truncated menin protein product. An in vitro protein truncation test (PTT) for diagnostic MEN1 screening proved to be too problematic for routine clinical laboratory implementation, given the problems inherent in (1) the instability of abnormal mRNA in peripheral blood lymphocytes of patients with MEN1 and (2) the scarcity of methionine residues for 35S-methionine substitution in labeling the amino terminus polypeptides of the menin protein. Newer approaches with nonisotopic PTT [25, 26] may be more successful in applying this strategy to MEN1 mutation detection. Identification of large deletions by Southern blot or fluorescence in situ hybridization analysis may be useful for mutation screening because deletion of an entire exon or exons will result in amplification and sequencing of the remaining, normal allele and a false-negative test result, although next-generation sequencing can detect large deletions. Genetic variants in the regulatory regions of MEN1, including promoter mutations or methylation defects, may indicate abnormal gene expression but have not yet been documented.
For the MEN1 patients with no demonstration of germline MEN1 gene mutation, biochemical screening is an alternative course for monitoring disease. Careful biochemical screening may reveal evidence of neoplasia, including hyperparathyroidism, 10 years prior to the expression of overt clinical symptoms [20].
Formal proficiency testing programs are not available for MEN1 molecular testing. Clinical laboratories must meet the proficiency testing requirements by alternative proficiency assessment methods.
Conclusions and Future Directions
Currently, no preventive measures are available for prophylaxis or cure of MEN1-associated tumors. Radiologic imaging for identification of MEN1-associated tumors generally has low sensitivity in asymptomatic individuals. Biochemical screening for the tumor-secreted hormones may help detect early disease in asymptomatic mutation carriers [27]. Treatment usually involves surgery to remove the tumors, sparing unaffected tissue to preserve normal hormone production. Several clinical trials of drug therapies for patients with MEN1 have been launched. Various approaches focus on the modulation of the hypercalcemia and hypercalciuria of primary hyperparathyroidism by administration of the calcium-sensing receptor agonist, cinacalcet [28], or high-dose somatostatin with octreotide-LAR [29]. More extensive studies are needed to establish the efficacy of these molecules for clinical use.
MEN2
Molecular Basis of Disease
In 1993, the RET proto-oncogene (“rearranged during transfection”), located on chromosome 10q11, was cloned and characterized as encoding a transmembrane tyrosine kinase with a cysteine-rich extracellular receptor domain (Fig. 25.2) present on neuroendocrine cells of neural crest origin [30, 31]. RET spans 60 kb with 21 exons encoding a protein of approximately 1,100 amino acids [1]. Glial cell-derived neurotrophic factor and neurturin are two ligands for the RET receptor domain [32]. The ligands initiate homodimerization of RET protein molecules resulting in phosphorylation and activation of the tyrosine kinase domain, leading to downstream signal transduction [32]. RET is the only gene known to be associated with MEN2. Missense mutations in the RET gene were characterized in individuals with the MEN2 syndromes [30, 31, 33, 34] with a very restricted pattern of variations (Fig. 25.2). In patients with MEN2A or FMTC, nearly all the mutations in this gene are activating, gain-of-function missense mutations localized to exons 10, 11, and 13–15. Most commonly, heterozygous missense mutations are found in a cysteine-rich, extracellular domain adjacent to the transmembrane domain of the RET protein including six conserved cysteine residues in RET exon 10 (codons 609, 611, 618, and 620) and exon 11 (codons 630 and 634) (Table 25.1 and Fig. 25.2) [30, 31, 33]. Most of these mutations replace a cysteine with another amino acid that cannot form intramolecular disulfide bonds. The unpaired cysteine is then able to form a disulfide bond with another RET protein molecule producing constitutive dimerization and increased kinase activation of the aberrant RET protein [35, 36]. Missense mutations in codons 768 and 804, located in exons 13 and 14, respectively (Table 25.1), affect the intracellular portions of the RET protein next to the transmembrane domain (Fig. 25.2) [37, 38]. Nearly 95 % of all patients diagnosed with MEN2B have been characterized with a single missense mutation in codon 918 of RET exon 16, M918T, replacing a methionine residue with a threonine [33, 34]. Another 2–3 % of patients have an Ala833Phe mutation in exon 15. Both variants affect the intracellular tyrosine kinase catalytic domain (Fig. 25.2), altering substrate recognition, which leads to cellular transformation [35]. In each case, these common RET mutations produce a gain-of-function change for the aberrant RET protein. Online information about many other RET mutations is available from the Human Gene Mutation Database Cardiff (http://www.hgmd.cf.ac.uk/ac/index.php, accessed 10/2/2014), the Weizmann Institute of Science GeneCards (http://www.genecards.org/cgi-bin/carddisp.pl?gene=RET&search=RET, accessed 10/2/2014), and the ARUP Institute for Clinical and Experimental Pathology (http://www.arup.utah.edu/database/MEN2/MEN2_welcome.php, accessed 10/2/2014).
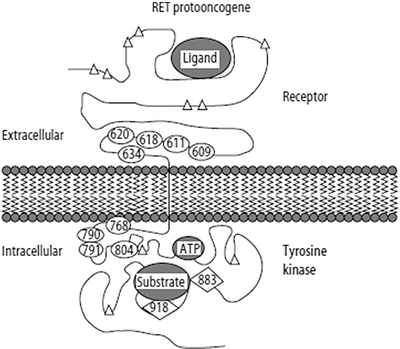
Figure 25.2
Diagram of RET gene product indicating the relative location of germline mutations characterized in patients with MEN2. Ovals are numbered to represent the codons that commonly contain germline missense mutations associated with MEN2A and FMTC. Diamonds are numbered to represent the codon location of germline mutations in MEN2B. Triangles indicate the positions of mutations in hereditary Hirschsprung disease. From Phay JE, Moley JF, Lairmore TC. Multiple endocrine neoplasias. Semin Surg Oncol 2000;18:324–32, copyright © 2000 [1]. Reprinted with permission from John Wiley and Sons
Table 25.1
Common RET gene mutations in MEN2 and associated clinical phenotypes
MEN2 phenotype b | |||||
|---|---|---|---|---|---|
RET location | Mutation a | FMTC | MEN2A | MEN2B | ATA risk c |
Exon 10 | C609Y, F, G, R, or S | Y | Y | N | B |
C611Y, F, G, R, S, or W | Y | Y | N | B | |
C618Y, F, G, R, or S | Y | Y | N | B | |
C620Y, F, G, R, S, or W | Y | Y | N | B | |
Exon 11 | C634R | N | Y | N | C |
C634Y, F, G, S, or W | Y | Y | N | C | |
Exon 13 | E768D | Y | Y | N | A |
Exon 14 | V804M or V804L | Y | Y | N | A |
V804M + E805K | N | N | Y | D | |
V804M + Y806C | N | N | Y | D | |
Exon 14/15 | V804M + S904C | N | N | Y | D |
Exon 15 | A883F | N | N | Y | D |
Exon 16 | M918 ATG | N | N | Y | D |
Hirschsprung disease (HSCR), an absence of enteric ganglia in the colon, also is associated with somatic or germline RET mutations, either in codons 609, 618, or 620, or distributed throughout other regions of the RET gene [40, 41]. Some families have been described in which HSCR cosegregates with either MEN2A or FMTC [42]. RET mutations in patients with HSCR may produce either loss or gain of RET protein function and include frameshift and nonsense mutations. Papillary thyroid carcinoma is associated with somatic gene rearrangements of the RET gene [43].
Clinical Utility of Testing
In an autosomal dominant inherited disorder, an individual with a single RET allele mutation has (1) a high likelihood of developing MTC (approximately 95 % of carriers) and (2) a 50 % risk of transmitting this risk allele and the high likelihood of developing MTC to each offspring. Genetic testing for RET mutations is the accepted best practice for confirmation of the diagnosis of MEN2, predictive risk-assessment testing for family members, and prenatal diagnosis of MEN2. Approximately 25 % of all MTC occurs as part of MEN2. Early detection of a pathogenic RET mutation improves the prognosis for presymptomatic individuals by offering an opportunity for therapeutic intervention prior to advanced disease, metastasis, or both [44–46]. Reduced morbidity and mortality is achieved by increased clinical monitoring, prophylactic thyroidectomy (followed by thyroid hormone replacement therapy plus autotransplantation of the parathyroids), or both [1, 47]. This can be a very effective treatment to prevent disease metastasis. Resected thyroid tissue from children and adults with positive genetic findings demonstrates C cell hyperplasia or microscopic foci of malignancy in the absence of biochemical screening abnormalities or clinical symptoms [46]. Thus, genetic testing for RET mutations is a more sensitive and specific screening tool than either physiological testing or histopathologic examination of the thyroid for assessing familial cancer risk for MEN2 in these families [44–46].
The clinical significance of identifying a RET mutation is considerable, given that there is virtually 100 % penetrance of these mutations for MTC. But genetic findings alone cannot predict the age of disease onset; thus, continued surveillance for residual or recurrent MTC plus adrenal tumors is included in the follow-up care of MEN2 patients and asymptomatic carriers. About 50 % of MEN2 patients (both subtypes A and B) will develop pheochromocytoma, while 20–30 % of MEN2A patients develop parathyroid hyperplasia [1]. Patients with FMTC typically develop only MTC and none of the other clinical manifestations of MEN2A or MEN2B. Each subtype appears to be consistent within a family. Any RET mutation in codon 634 in exon 11 results in a higher incidence of pheochromocytomas and hyperparathyroidism. In particular, the C634R (Cys to Arg) mutation has been associated with a higher incidence of metastases at diagnosis; the allele is virtually absent in patients with FMTC [48]. Families with both MEN2A or FMTC and HRSC most frequently have germline RET mutations in codons 609, 618, or 620 in exon 10 [49]. Individuals within the same family who carry the same RET mutation may still display a variable phenotype indicating that additional modifier gene functions or environmental or epigenetic factors affect the expression of altered RET protein function and tumorigenesis. Concomitant mutations in genes coding for RET ligands or coreceptors are the most likely modifier molecules.
Genetic screening and mutation identification are currently recommended by 5 years of age for children who are at risk in MEN2A families. Children should be screened at birth if MEN2B has been diagnosed in close relatives, due to the earlier age of onset and aggressive clinical course of the MEN2B type [44, 45]. Patients with MEN2B should be treated as soon as the diagnosis is made. Some of the other advantages of DNA testing in MEN2 are that the test is relatively noninvasive and low risk, it is usually better tolerated by the patient than biochemical screening by metabolic challenge [47], genotype results are not subject to physiologic status, and serial genotype testing is not necessary. Individuals in MEN2 families who are not carriers of the familial RET mutation have the lower, general population risk for sporadic incidence of these endocrine neoplasias and do not require the same frequent monitoring for abnormal thyroid or adrenal function in the absence of clinical symptoms.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


