Figure 2.1
The polymerase chain reaction
The hybridized PCR primers form local areas of double strandedness with the template DNA, thereby serving as sites for DNA polymerase to bind and synthesize a new strand of DNA, using the target DNA as a template and dNTPS present in the reaction solution. Subsequent to the initial discovery of PCR, the opportunity for automating the temperature cycling was realized by using DNA polymerase from hot-spring living bacteria, Thermus aquaticus (hence the term “Taq polymerase”). T. aquaticus thrives at very high temperatures, and so its proteins do not denature at the high temperatures needed to denature DNA in the first step of PCR. Catalysis by Taq polymerase of a new strand of DNA proceeds at a temperature intermediate to the near-boiling temperature used for denaturation and the relatively lower temperature used for annealing. DNA polymerization occurs during this extension step, typically at 65–75 °C. Taken together, these three steps (denaturation, annealing, and extension) define one PCR cycle.
Temperature cycling is automated through the use of an instrument called a thermal cycler. Thermal cyclers hold small capped tubes (or 96- or 384-well microtiter plates for larger volume testing) containing the reagents needed for PCR, and cycle among the temperatures needed for the different steps of the PCR [11]. A single PCR tube contains template DNA (<1 ng to 1 μg), Taq DNA polymerase, two PCR primers (15–30 nucleotides long), all four dNTPs, Mg2+, and buffer to maintain an elevated pH (8.4) optimal for Taq polymerase activity.
The repetition of the cycles generates exponential amplification of the target DNA because each double-stranded target DNA molecule, theoretically even if there is only one, is replicated after one PCR cycle. Both the original and replicated DNA molecules can function as templates for cycle 2, in true “chain reaction” style, generating another doubling, or four copies of the original target. Cycle 3 ends with eight molecules, and doubling continues with completion of each new cycle. This doubling plateaus in later cycles since reagents, usually dNTPs, become limiting. Additionally, the enzyme may not function at 100 % efficiency, and so true exponential amplification is theoretical, although there is a true exponential phase of amplification.
Greater than one billion copies of the original target DNA region are generated after 32 cycles of PCR: 232 or more than four billion, the difference owing to the fact that unit-length amplicons are not generated until the end of the second cycle of PCR. Amplicons (PCR products) are defined as replicated target molecules created by PCR. Unit-length amplicons are those whose ends are defined by the primers. During the first cycle, the primers are extended by Taq polymerase using template DNA. The termination of this extension is undefined and a function of how far the polymerase moves down the template during the time allotted by the temperature cycle. The enzyme, therefore, moves beyond the ends of the primer-binding site on the complementary strand. After completion of the first cycle, therefore, the newly synthesized DNA molecules are greater in length than the sequence bracketed on each strand by the primers. In the second cycle, DNA molecules are synthesized from the products of the first cycle whose ends are defined by the two primers, thus generating unit-length or specific amplicons. While all of the above is true, the practical clinical laboratory difference between one- and four billion-fold amplification is irrelevant because either number is sufficient for detection of the target, e.g., by electrophoresis with SYBR green or EtBr used as an intercalating agent for visualization.
Several factors affect PCR specificity and sensitivity. The production of specific PCR amplicons is a function of both the complementarity of the primers to the target DNA and the annealing temperature of the PCR cycle. Heating denatures the primer and target DNA. The temperature at which a primer melts from the target DNA varies directly with the length of the primer and the guanine–cytosine (GC) content of the primer, and inversely with the degree of mismatch between the primer and the target DNA. The melting temperature (T m) of the primer is the temperature at which 50 % of the primer is denatured from the target DNA. If the thermal cycler is programmed to reach an annealing temperature higher than the primer T m, the efficiency of PCR is compromised and sensitivity decreased. In contrast, if the annealing temperature is substantially less than the primer T m, the primer can bind to both complementary and noncomplementary DNA, resulting in reduced PCR specificity as nontarget DNA is amplified (and potentially decreased sensitivity as reaction components are used nonspecifically). Therefore, the ideal annealing temperature is slightly less than the Tm of both primers, and the primers should be designed to have a very similar T m. The annealing temperature can be decreased with subsequent cycles during PCR in a process called “touchdown” PCR. This allows the initial cycles to produce specific products at high annealing temperatures, while later cycles amplify previously generated amplicons more efficiently using lower annealing temperatures, thereby increasing sensitivity (see below the use of touchdown PCR in multiplex PCR).
Taq polymerase is very sensitive to mismatches between the primer and the target DNA at the 3′ end of the primer but can withstand considerable noncomplementarity at the 5′ end of the primer. Numerous PCR variations have been designed to take advantage of both these facts. Taq polymerase also requires Mg2+ as a cofactor for stabilization of primer annealing. Insufficient Mg2+ decreases PCR efficiency, while too much Mg2+ stabilizes nonspecific primer annealing. Primers with a high GC content may show a narrow range of tolerance for variation from ideal PCR conditions, leading to decreased amplification or nonspecific products. This may be alleviated by using PCR additives such as dimethyl sulfoxide (DMSO), betaine or glycerol, but the success and amount of these additives may need to be determined empirically for different primer pairs. Another strategy to improve specificity is the use of “hot-start” PCR, in which a crucial PCR reactant such as Taq is either physically or chemically sequestered from other PCR reagents until denaturation begins. This prevents the generation of nonspecific amplification products by inhibiting the activity of Taq at lower temperatures and until after the initial PCR denaturation step.
PCR is more sensitive than Southern blot hybridization because of the amplification of the target sequence. However, the specificity of the amplified PCR product(s) must be verified. Simple agarose gel electrophoresis coupled with intercalating agent staining may be used to observe the PCR product(s). When a clinical PCR protocol is established, such gels may be subjected the first time to blot hybridization with a specific probe complementary to the internal, non-primer sequence of the amplicon(s). This exercise proves that the PCR-generated band not only is the correct size and highly likely to be the correct target, but also is a DNA fragment that has high or perfect homology with a known probe or the correct target sequence. For example, hybridization of a particular 302 bp PCR product band detectable on an agarose gel with a defined cytomegalovirus (CMV) DNA probe confirms that the oligonucleotide primers synthesized based on the CMV sequence and used in the PCR are recognizing CMV-specific DNA and that the PCR is indeed specific for CMV sequence. An alternative method to validate the specificity of the PCR product is to sequence the PCR product. Following this one-time validation analysis, electrophoresis alone may be the assay endpoint, as opposed to blot hybridization or sequencing.
There have been significant commercial endeavors to automate or semiautomate high-volume PCR-based clinical tests. For example, denatured aliquots of completed PCRs can be added to microtiter plates with wells to which specific DNA probes are bound. In the presence of amplicon, if the patient is infected with the pathogen of interest or a specific mutation is present, the amplicons hybridize to the bound probe and are retained in the well during subsequent washing steps. Biochemical reactions are used to detect labeled moieties in the amplicons (“built in” to the PCR components), facilitating colorimetric detection of a positive patient reaction by an automated plate reader. Absence of colored product in a well indicates a negative result for that patient specimen, provided that all positive and negative controls are within tolerance limits. This scheme has gained US Food and Drug Administration (FDA) approval or clearance for clinical PCR-based detection kits for Chlamydia trachomatis, Neisseria gonorrhoeae, HCV (qualitative), and HIV [12]. (For a complete list of FDA-approved or -cleared tests, go to http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm). Subsequent generations of PCR instrumentation are available that completely automate the amplification and detection processes [13].
Another aspect of PCR that is attractive for the clinical molecular laboratory is the ability to use relatively crude DNA extractions from patient specimens rather than highly purified DNA. Cell lysis and subsequent DNA liberation accomplished by boiling or treatment with detergent may be sufficient to process a specimen in preparation for PCR [14]. Conventional PCR-based tests may be completed with turnaround times of as short as 2–4 h, while real-time PCR can be completed in 30 min, making this technique attractive for rapid clinical testing.
Examples of Applications of PCR
1.
Detection of the diagnostic BCL2–IGH gene rearrangement in follicular lymphoma
2.
Detection of Chlamydia trachomatis in urine
PCR Variations
PCR-Restriction Fragment Length Polymorphism Analysis
Polymorphisms are inherited differences found among the individuals in a population at a frequency >1 % of that population. The term “polymorphism” is not synonymous with the term “mutation” which is used for germline variations that are pathogenic and found less frequently in a population, or are nongermline changes in a tumor cell (somatic mutations). In the case of restriction fragment length polymorphisms (RFLP), DNA sequence differences alter RE recognition sites, manifested either as obliteration or creation of a restriction site. With obliteration of a RE site, the DNA of individuals with an RFLP exhibits a larger restriction fragment of DNA than those without the polymorphism. With creation of a new RE site, RE digestion results in two smaller fragments relative to the individual without the polymorphism. In either case, the polymorphism is detectable by creation of a new restriction fragment pattern, that is, a restriction fragment length polymorphism. In PCR-RFLP, the PCR products are digested by one or a combination of REs and electrophoresed to detect polymorphisms or mutations which are seen as changes in the DNA fragment sizes reflected by changes in the band pattern on the gel (or chromatogram).
Examples of Applications of PCR-RFLP Analysis
1.
Detection of sickle-cell hemoglobin (HbS) gene mutation
2.
Detection of the MnlI restriction enzyme polymorphism created by the Factor V Leiden mutation [15]
Restriction-Site Generating PCR
Some DNA sequence variants create or abolish RE recognition sites and can easily be detected by PCR-RFLP. Unfortunately, most variants do not alter a RE recognition site. In restriction-site generating PCR (RG-PCR) (and a related research technique called PCR-mediated site-directed mutagenesis [PSDM]), an artificial RE recognition site is generated during PCR using a specially designed PCR primer [16, 17]. The primer contains a base mismatch to the template DNA adjacent to the variable base of the variant that creates a RE recognition site in the PCR product. The mismatched base in the primer is located near or at the 3′ end of the primer, which is near or adjacent to the variable base of the variant, and together they create a novel restriction site within either the variant or non-variant amplicon. The presence or absence of the RE recognition site is determined from the pattern of digested PCR product fragments by gel electrophoresis. Not all sequences are amenable to the generation of a restriction site, and the amplification efficiency is often decreased due to destabilization of the primer with the mismatch.
Examples of Applications of RG-PCR
1.
Identification of mutations in the CTFR gene in cystic fibrosis
2.
Identification of mutations in the ATM gene in ataxia–telangiectasis
Multiplex PCR
Multiplex PCR is a technique used for amplification of several discrete genetic loci with multiple PCR primer pairs in a single reaction. Multiplex PCR simultaneously answers several related questions about a specimen without the need for multiple individual PCR reactions. Multiplex PCR is commonly used for verification that amplifiable nucleic acid is present in the sample, for example, amplification of a housekeeping gene in addition to the gene sequence(s) of interest, and to check for the presence of PCR inhibitors that can prevent amplification of target nucleic acid, for example, coamplification of an exogenously added internal control. Multiplex PCR often requires painstaking optimization of PCR conditions and careful design of the multiple primer pairs to match PCR efficiencies and to prevent the generation of primer-dimers (PCR products generated by the primers alone due to complementarity between primer regions) and other nonspecific PCR products that may interfere with the amplification of specific products. Touchdown PCR can be used with multiplex PCR if the primer pairs have different annealing temperatures. Concentrations of individual primer pairs may need to be optimized to account for different amplification efficiencies and competition between the primer pairs.
Examples of Applications of Multiplex PCR
1.
Detection of enterovirus and herpes simplex virus (HSV) nucleic acids in cerebrospinal fluid (CSF)
2.
Detection of pathogenic enteric bacteria in stool
5.
Amplification of multiple microsatellite loci for bone marrow engraftment analysis
Single Nucleotide Extension
Another method for a multiplexed assay is single nucleotide extension (SNE) or single base extension (SBE). In this method, either a single long-range PCR or a multiplexed PCR is used to amplify the region(s) of interest. This is followed by a multiplexed set of extension primers of differing lengths that hybridize one base upstream to the variant(s) of interest. A second, linear amplification, similar to Sanger sequencing, adds the next nucleotide (at the variant position) using ddNTPs, with each type labeled with a different fluorophore. The products are separated by CE or mass spectrometry, and the specific fluorescent signal of the incorporated base indicates which base was added, and whether the variant is present or not. This method can be used to genotype up to approximately 20 mutations at once. SNE and SBE can be considered sequencing, but of just one base.
Examples of Applications of SNE
Nested PCR
For nested PCR, two pairs of PCR primers with one set internal to the other (nested) are used to sequentially amplify a single locus. The first pair is used to amplify the locus as in any PCR assay. A dilution of the first PCR reaction then is amplified with the nested primers. Alternatively, semi-nested PCR is performed using one of the original PCR primers and one new internal primer in a second round of amplification. Both nested and semi-nested PCR generate a second PCR product that is shorter than the first one [22]. The logic behind this strategy is that if the wrong locus was amplified incorrectly or nonspecifically, the probability is very low that it would be amplified a second time by a second pair of primers. Thus, nested PCR enhances specificity while also increasing sensitivity. The problem with nested PCR is the high risk of amplicon contamination when the first-round PCR products are used to set up the second round of PCR with the nested primers (see Amplicon Carryover Contamination section below for information on PCR contamination control). For this reason, many clinical laboratories do not use nested PCR procedures.
Allele-Specific PCR
Allele-specific PCR (AS-PCR) also is referred to as amplification refractory mutation system (ARMS), PCR amplification of specific alleles (PASA) and PCR amplification with sequence-specific primers (PCR-SSP). AS-PCR is based on the principle that a 3′ mismatch between a PCR primer and the template DNA prevents PCR amplification [23]. AS-PCR is especially useful for detection of single nucleotide polymorphisms (SNPs) or mutations. For AS-PCR, target DNA is amplified in two separate and simultaneous reactions. Each reaction contains an allele-specific primer (either non-variant or variant) and a second primer common to both reactions. PCR is performed under stringent conditions, to prevent PCR amplification if a mismatch is present. Genotype is based on amplification in either one of the reactions alone (homozygous non-variant or variant) or both reactions (heterozygous). Detection of the amplicon is either by gel electrophoresis or real-time PCR technology (see below). A disadvantage of AS-PCR is that unsuspected nucleotide variants located in the DNA template at or adjacent to the 3′ binding site of the primer would prevent amplification, leading to incorrect genotyping.
AS-PCR can detect one variant allele in the presence of 40 copies of the non-variant allele. In addition, AS-PCR can be combined with multiplex PCR using multiple allele-specific primers in the same reaction tube. This technique is known as multiplex ARMS, a useful method when a single disease is caused by different mutations in one or more genes. Multiplex PCR-SSP also is commonly used in low-resolution HLA typing, in which multiple primer pairs for HLA loci are used along with control primers that amplify a housekeeping gene to verify that amplifiable DNA is present in each reaction tube.
Examples of Applications of AS-PCR
1.
Detection of multiple cystic fibrosis CFTR mutations
2.
Detection of common α-1 antitrypsin deficiency mutations
3.
Detection of common phenylketonuria mutations
Allele-Specific Oligonucleotide Hybridization
Allele-specific oligonucleotide hybridization (ASOH), also known as dot-blot analysis, is used for genotyping of highly polymorphic regions of DNA. ASOH can be thought of as a variation of the Southern blot, in that patient DNA amplified by PCR is bound to a membrane and hybridized with labeled allele-specific oligonucleotide probes [24]. Reverse dot-blot analysis differs from ASOH in that unlabeled allele-specific oligonucleotide probes are spotted onto different membrane locations and hybridized with labeled PCR amplicons.
For ASOH, the PCR products are denatured and a small amount of denatured (single stranded) amplicon is spotted onto a nylon or nitrocellulose membrane. The amplicon is permanently bound to the membrane by baking under vacuum or UV cross-linking. Amplicons from different specimens can be spotted at different locations to interrogate the genotype of multiple specimens simultaneously. Duplicate membranes are made for each probe type. Each membrane is hybridized with two different labeled oligonucleotide probes (one complementary to the variant sequence and another to the non-variant sequence of the same DNA region). The membranes are washed to remove nonspecifically bound probe. Samples that hybridize strongly to only one probe indicate homozygosity for the non-variant or variant allele; those that hybridize with both probes are heterozygous. The oligonucleotide probes are labeled and detected by radioactivity (often avoided in clinical molecular laboratories), fluorescence, colorimetry, chemiluminescence or mass spectrometry. One drawback of ASOH is the potentially ambiguous discrimination of a positive compared to a negative signal. Optimization of the assay and the use of both positive and negative controls help to define and score ASOH results.
Example of Application of ASOH
1.
Low-resolution HLA typing
Oligonucleotide Ligation Assay
Oligonucleotide ligation assay (OLA) is a highly specific method for detecting well-defined alleles that differ by a single base [25, 26]. The target sequence is initially amplified using PCR and then denatured. A pair of allele-specific oligonucleotide (ASO) probes (one specific for the non-variant allele and the other specific for the variant allele), a common reporter probe (complementary to a sequence common to both alleles), and DNA ligase are added to the denatured PCR products. The ASO probes are designed to differ from one another only at the terminal 3′ base. The common reporter probe is positioned immediately adjacent to the 3′ terminal end of the ASO probes. If the ASO is complementary to the amplicon, DNA ligase can covalently join the adjacent ASO and reporter probes. If the ASO is not a perfect match to the amplicon, the 3′ base does not anneal with the template DNA, and DNA ligase cannot join the ASO and reporter probes. The ligation products are analyzed by electrophoresis. Alternatively, one of the probes can be biotinylated at the 5′ end and the other probe tagged at the 3′ end with a reporter molecule such as fluorescein or digoxigenin. If ligation occurs, the ligation product is biotinylated at one end, facilitating capture onto a streptavidin-coated microtiter plate. The opposite end contains the reporter label. Washing removes unbound label and the reporter molecule is detected.
Example of Application of OLA
1.
Detection of multiple CFTR mutations for cystic fibrosis
High-Resolution Melting Curve Analysis
Melting curve analysis takes advantage of the principle that DNA sequences that are a perfect match will melt at a higher temperature than those that contain either a heterozygous or homozygous nucleotide variant. Typically, in genotyping by high-resolution melting curve analysis, an area of interest (approximately 50 bases, including primers) is amplified in the presence of a double-stranded DNA-intercalating fluorophore or double-stranded DNA-intercalating dye. After amplification, the temperature is decreased to the point that the DNA will reanneal. The temperature then gradually is increased while the fluorescence is monitored. Variants are identified by a change in melting curve shape as compared to a non-variant control. While the typical use of melting curve analysis is to identify single nucleotide variants of interest, it can also be used as a rapid scanning method to detect potential sequence variants in a gene of interest. Melting curve analysis can be affected by factors such as salt concentration and DNA quantity, so all samples and controls must be prepared and amplified in an identical manner to exclude this as a possible confounding factor.
Examples of Applications of Melting Curve Analysis
1.
Factor V Leiden genotyping
2.
HFE-associated hereditary hemochromatosis genotyping
Pyrosequencing
Pyrosequencing is a useful method for variant detection when analytical sensitivity (limit of detection) or quantitation is important. In pyrosequencing, amplified targets are sequenced by adding and detecting incorporation of nucleotides one at a time. First, a target region is amplified and PCR products are captured through use of a biotinylated primer, which has been included in the PCR, along with a streptavidin-coated bead. Capture of the product onto the bead via the incorporated biotin group allows purification of the specific PCR product, followed by denaturation to create a single-stranded target. A sequencing primer close to the region of interest is then annealed to the captured single-stranded DNA amplicon. Deoxynucleotides are added one at a time in the presence of four enzymes: polymerase, sulfurylase, luciferase, and apyrase. Incorporation of the nucleotide releases pyrophosphate which participates in a chain reaction with luciferin, facilitated by sulfurylase and luciferase, to generate light. The amount of light released is directly proportional to the quantity of nucleotide incorporated. Apyrase removes unincorporated nucleotides. If the complementary base is not on the strand being sequenced, then no incorporation occurs and no light is released. The next nucleotide is then added to the pyrosequencing reaction and the steps are repeated. Nucleotides may be added in cyclic fashion (ACGTACGT…) or in an order specific to the target sequence, with allowance for anticipated variants.
Compared to other methodologies, pyrosequencing is particularly useful when analytical sensitivity is of particular concern, such as in detection of somatic mutations in tumor specimens which yield both non-variant and variant DNA. Analytical sensitivity of 5 % can be achieved with pyrosequencing, as compared to approximately 20 % for Sanger sequencing and approximately 10 % for melting curve analysis [27]. Quantification of mutant alleles also is provided by pyrosequencing results. Pyrosequencing is best suited for detection of variants within a targeted region. As compared to scanning methodologies, the region of interrogation may be somewhat smaller (under 100 bases, typically just multiple codons), but variants are both detected and characterized. An example of pyrosequencing results for codon 600 of BRAF is shown in Fig. 2.2.
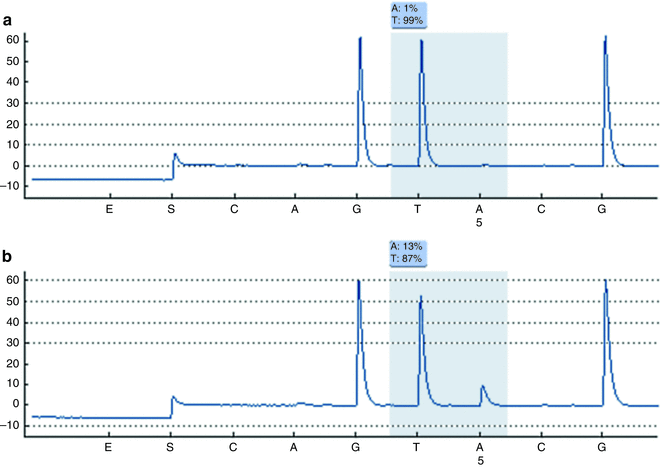
Figure 2.2
Pyrosequencing graphs for BRAF codon 600. Nucleotides were dispensed in the following order: CAGTACG. (a) Results for a non-variant sample showing the sequence GTG for codon 600. (b) Results for a variant sample (V600E), which has alleles with the sequence GAG, in addition to alleles harboring the normal sequence. Variant (V600E) alleles are present in this sample at 13 %.
Examples of Applications of Pyrosequencing
1.
KRAS mutation detection in multiple tumor types
2.
BRAF mutation detection in multiple tumor types
3.
LINE-1 methylation
Reverse Transcription-Polymerase Chain Reaction
Reverse transcription-polymerase chain reaction (RT-PCR) may be thought of as RNA-based PCR. RT-PCR was made possible by the discovery in the early 1970s of retroviral reverse transcriptase (RT), an RNA-dependent DNA polymerase, by Baltimore and Temin [28], for which they shared the Nobel Prize in 1975. Reverse transcriptase catalyzes DNA synthesis using RNA as the template, producing a DNA strand complementary to the RNA template, called complementary DNA (cDNA). Complementary DNA is far more stable than the corresponding RNA because it is not subject to degradation by RNase. Complementary DNA can be treated like any other DNA target in subsequent PCR. Logistically, RT-PCR is trivially more time-consuming than PCR due to the extra enzymatic step of reverse transcription, but there are enzymes that combine reverse transcription and DNA polymerase activities, facilitating the use of RT-PCR in the clinical molecular laboratory. With the introduction of techniques to successfully isolate and protect RNA from ubiquitous RNases, to synthesize cDNA by reverse transcription and with the discovery of PCR, RNA analysis is virtually as rapid and sensitive as PCR-based DNA investigation. RT-PCR is a high-volume test method for the clinical molecular laboratory as used for the diagnosis and quantification of RNA viruses in human specimens, principally HIV and HCV.
Examples of Applications of RT-PCR
1.
HIV and HCV viral load determinations
2.
Detection of BCR–ABL translocation diagnostic of chronic myelogenous leukemia
Real-Time (Quantitative) PCR
Real-time (quantitative) PCR is based on the generation of a fluorescent signal by the PCR process, which is detected during PCR cycling, i.e., in real time, and reflects the amount of PCR product synthesized [29–31]. Different real-time PCR methods use alternative ways to generate a fluorescent signal during PCR. These include an intercalating dye such as SYBR Green that binds the minor groove of DNA, or an oligonucleotide used as a primer or probe and labeled with a fluorogenic dye. Instruments that combine in vitro nucleic acid amplification and real-time detection of the PCR product dramatically increased testing options for oncology, infectious diseases and genetics because of the wide range of readily available amplification primers and detection schemes, rapid turnaround time, and reduced risk of PCR amplicon contamination.
Real-time PCR is different from conventional PCR in several ways. Amplicon generation, temperature profiles and melting curves are monitored in real time, reducing the time required for post-PCR analysis. In most applications, postamplification processing of the PCR products by gel electrophoresis or other method is eliminated. Because the reaction tubes remain closed after PCR starts, risk of amplicon carryover contamination within the laboratory is reduced. Results are more reproducible between runs since quantitation of target is based on amplification cycle threshold in the log-linear phase of amplification rather than traditional endpoint analysis in the PCR plateau phase. Real-time PCR methods have a wide dynamic range, up to 10 logs. Real-time PCR systems with intercalating dye or fluorogenic probes can be used to perform melting curve analysis, adding a check for the specificity of amplification or potentially the detection of unknown sequence variants (see below).
The simplest real-time PCR method uses intercalating dyes that insert into the stacked bases of DNA PCR products, allowing detection of amplification in real time. These dyes, for example, SYBR Green and ethidium bromide (EtBr), are nonsequence-specific dyes that increase in fluorescence when bound to double-stranded DNA. Intercalating dyes are used for melting curve analysis, qualitative and semiquantitative PCR, product discrimination and purity, and determination of primer and probe melting T m. Intercalating dyes can be used for quantitative PCR. Results, however, are more specific and accurate with a sequence-specific probe for real time monitoring of amplicon production since fluorescence is directly proportional to the amount of specific amplicon produced which reduces the background contributed by primer-dimers or nonspecific PCR products. Intercalating dye fluorescence represents all double-stranded DNA, including primer-dimers and other nonspecific products that can be visualized with an endpoint melting curve analysis.
Most fluorogenic oligonucleotide techniques take advantage of the principle of fluorescent resonance energy transfer (FRET), in which the energy from one dye molecule (the donor) is transferred without the emission of a photon to an acceptor dye molecule when the two are in close proximity. If the acceptor is a fluorophore, a photon is emitted at a characteristic wavelength. However, if the acceptor does not emit a photon, the energy is dissipated and fluorescence from the donor is quenched. The reporter dye can be either the donor (if no FRET takes place) or the acceptor (if FRET does take place) and is defined as the one whose fluorescence correlates with the quantity of desired PCR amplicon. Several fluorogenic techniques are described below.
TaqMan
The TaqMan technique uses a short probe complementary to a non-primer internal sequence of the PCR product. The probe is labeled at the 5′ end with a reporter donor dye and at the 3′ end with an acceptor dye that quenches the reporter when the probe is intact. During the extension phase of PCR, probe bound to an amplicon is cleaved by the 5′ endonuclease activity of Taq polymerase, freeing the reporter dye from the quencher and resulting in fluorescence. The fluorescent signal increases proportionally to the number of amplicons generated during the log-linear phase of amplification. To ensure that hydrolysis of the probe occurs, a two-step PCR can be used with annealing and extension taking place at the same temperature (approximately 60 °C). Ideally, the TaqMan probe binding site is located near one primer and the size of the amplicon is no longer than 200–300 bases. One negative aspect of this method is that once the probe is hydrolyzed, it is unavailable for subsequent reactions or melting curve analysis, thus requiring an excess amount of probe in the reaction mix with the potential to decrease the PCR efficiency.
Molecular Beacon
A molecular beacon is a probe with a 5′ reporter dye and 3′ quencher dye, which forms a hairpin loop structure when not bound to target DNA, thereby juxtaposing the reporter and quencher dyes with quenching of fluorescence. The loop sequence is complementary to the non-primer amplicon sequence. When the loop of the molecular beacon probe hybridizes to the amplicon during the annealing step of real-time PCR, the reporter dye is separated from the quencher, resulting in fluorescence. For the molecular beacon probe to anneal to the amplicon, the amplicon-probe hybrid must be more stable than the internal base-pairing stem of the hairpin so that a fluorescent signal is generated. Generally, DABCYL is the nonfluorescent universal quencher and the other dye is a reporter fluorophore such as FAM, Cy3, TET, TAMRA, Texas Red, ROX, or Cy5.
Hybridization
Hybridization is typically a two-probe system in which one probe contains a donor dye and the other contains the reporter acceptor dye. The probes are designed to anneal to one strand of the PCR product adjacent to one another and internal to the primers. This juxtaposes the dyes, allowing FRET to occur. This probe format works well with the traditional three-step PCR with annealing at approximately 55 °C (primer specific) and extension at 72 °C, the optimal temperature for Taq polymerase activity. When DNA polymerase encounters the probes, they are displaced from the target strand rather than hydrolyzed and thus are available for the next round of amplification as well as endpoint melting curve analysis. In a variation of this method, a single unlabeled probe may be used in conjunction with an intercalating dye.
Uniprimer (Amplifluor, Sunrise)
Like molecular beacon probes, the uniprimer system uses a hairpin structure in the probe to quench fluorescence. The 3′ region of the fluorogenic probe is identical to a nonbinding region at the 5′ end of the reverse PCR primer. This allows the fluorogenic probe to become a primer for the newly formed amplicon by the third round of PCR. The probe is then opened in the fourth and subsequent rounds of PCR by the polymerase action of Taq, allowing fluorescence to occur. The advantage of this system is that the same fluorogenic probe sequence can be used in any PCR reaction (universal fluorogenic primer).
Scorpion
Scorpion also uses a hairpin structure in the probe to quench fluorescence. The fluorogenic probe is part of the reverse primer, and the nucleotides in the hairpin are complementary to the PCR amplicon sequence between the primers. The Scorpion probe unfolds and anneals to the PCR amplicon, allowing fluorescence to take place beginning in the first round of PCR.
Lux
Lux is a variation of real-time PCR that uses a single fluorophore in a primer with a hairpin loop structure. The fluorophore is quenched by the complementary structure of nucleotides in the stem of the hairpin. When the primer is incorporated into double-stranded DNA, thus opening the hairpin, fluorescence is maximal. The advantage of this system is lower production costs with the use of only one fluorophore.
The following concepts are important for understanding the use of real-time PCR in a clinical molecular laboratory. When optimizing real-time PCR, the amplification curve of the fluorescent signal vs the number of PCR cycles should be monitored to determine when optimal conditions have been achieved. The amplification curve should be sigmoidal (S shaped) with three phases: baseline (background signal or lag phase), log-linear (exponential amplification phase), and plateau. For each phase, several characteristics should be assessed. The baseline phase of the curve represents initial cycles of amplification in which accumulation of the specific signal has not yet exceeded the background signal. The fluorescent signal in this phase is from unbound probe or autofluorescing components in the reaction. The log-linear phase of the curve represents exponential amplification of the target and provides useful information about the reaction. The curve can be described by the following equation: T n = T 0(E) n , where T n is the amount of target sequence at cycle n, T 0 is the initial amount of target sequence at cycle 0, and E is the amplification efficiency of the target sequence. The crossing point represents the number of PCR cycles at which the amplification curve enters the log-linear phase. There is an inverse linear relationship between the crossing-point cycle number and the number of template copies present in a reaction.
The slope of the log-linear phase is a reflection of amplification efficiency, and the efficiency of the reaction can be determined by identifying the crossing points of known standards and plotting a line of linear regression (Fig. 2.3). The efficiency can then be determined using the following equation: E = 10−1/slope, where E is efficiency and slope is the slope of the standard curve. Using this equation, the slope should be between -3 and -4, with -3.3 indicative of efficiency close to or at 2. The inflection point of the amplification curve is the point at which the log-linear amplification curve goes from positive to negative and begins to enter the plateau phase. If there is no inflection point, the curve may represent not amplification of DNA, but rather signal drift. Drift is characterized by gradual increase or decrease in fluorescence without amplification of product.
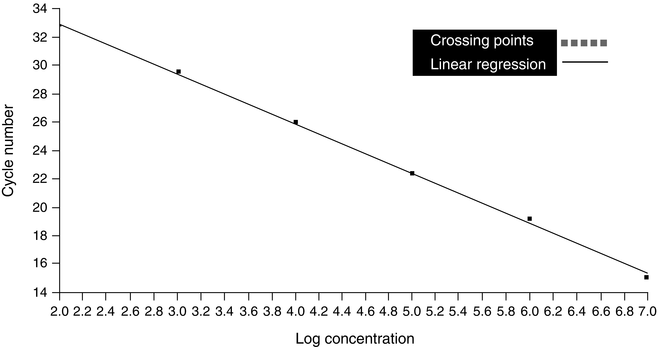
Figure 2.3
Standard curve generated from results of real-time PCR of a tenfold dilution series of a known standard
Plateau is defined as the phase of amplification when critical components of the PCR become rate limiting and amplicon accumulation is minimized or stops. The plateau is also the point at which incremental increase in fluorescent signal stops. As the rate of accumulation slows and enters the plateau phase, the curve levels. Since endpoint measurements often are made in conventional PCR when reaction components are limited, minor sample variations can have a relatively major effect on endpoint product production. The plateau phase can be shortened by decreasing the number of PCR cycles for reduced nonspecific amplicon production. Several factors contribute to the plateau phase: PCR product reannealing vs primer annealing, enzyme or dNTPs becoming limiting, and amplicon buildup with resultant reaction inhibition.
In real-time PCR, the log-linear phase of the amplification curve is used for data analysis and provides a more accurate measurement than endpoint analysis. The cycle at which the curve crosses a specified threshold is called the cycle threshold (Ct), or crossing point (Cp). The Ct value can be used for qualitative or quantitative analysis. A qualitative analysis uses the defined Ct as a pass/fail measurement. A quantitative assay uses the Ct of defined standards of known template concentration to generate a standard curve. Then, the Ct values for unknown samples are used to extrapolate the concentration(s) in the unknown samples from the standard curve. Some commercial real-time PCR software allows determination of the Ct by a mathematical analysis of the amplification curve, rather than crossing at a set fluorescent signal threshold. Plotting the second derivative of the amplification curve generates a peak that corresponds to a point near the baseline of the growth curve (see Fig. 2.4). The cycle at which this peak occurs is designated as the Ct or Cp. This analysis method can provide better run-to-run reproducibility than manually setting the Ct using the primary signal.
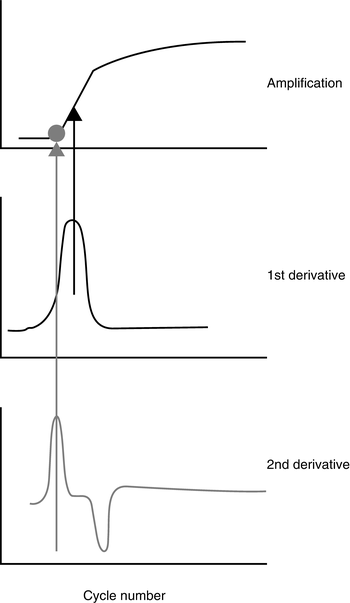
Figure 2.4
Graphical depiction of the second derivative maximum method used to identify the crossing point (Y axis is fluorescence value)
DNA Methylation and Methylation-Specific PCR
DNA methylation is a mechanism by which the cell regulates gene expression. Methylation is an enzyme-mediated modification that adds a methyl (-CH3) group at a selected site on DNA or RNA. In humans, methylation occurs only at cytosine (C) bases adjacent to a guanine (G) base, known as CpG dinucleotides. CpG dinucleotides are prone to spontaneous mutation and have been selectively depleted from the mammalian genome. However, some regions of DNA contain CpG dinucleotides and are referred to as CpG islands. CpG islands are found primarily in the 5′ region of expressed genes, often in association with promoters. When the promoter CpG island is methylated, the corresponding gene is silenced and transcription does not occur. This is one method of silencing imprinted genes, as the methylation pattern and resulting transcription repression is passed on through cell divisions. Aberrant CpG island methylation of tumor-suppressor genes is frequent in cancer and appears to be an important mechanism of neoplastic transformation.
Methylated DNA can be distinguished from unmethylated DNA using sodium bisulfite treatment of DNA, which converts unmethylated C to uracil (U) but leaves methylated C intact [32]. This in vitro treatment can be assessed by one of several methods to distinguish C from U, including restriction endonuclease digestion with methylation-sensitive enzymes, sequencing, or methylation-specific PCR (MSP) [33]. In MSP of bisulfite-treated DNA, primer pairs that specifically identify either methylated or unmethylated DNA are used. The primers are designed to hybridize to regions containing one to three CpG sites concentrated in the 3′ region of the primer to increase amplification specificity, and enough non-CpG cytosines to ensure that unmodified DNA is not amplified. Gel electrophoresis is used to detect the presence or absence of the amplicon in each of the two reactions, indicating the presence of unmethylated or methylated alleles or both. A modification of quantitative MSP combines MSP with real-time PCR to distinguish the high-level CpG methylation in neoplasia from low-level methylation that can occur with aging or in nonneoplastic conditions such as metaplasia [34].
Examples of Applications of Methylation-Specific PCR
1.
Analysis of imprinted genes in Prader–Willi and Angelman Syndromes
2.
Clonality assessment based on X chromosome inactivation
3.
Abnormal methylation in neoplasia
Mass Spectrometry
Mass spectrometry (MS) is a flexible platform for variant and target detection for clinical laboratory applications. In preparation for MS-based detection, a variety of PCR methods such as SNE can be adopted. PCR is performed to amplify the region(s) of interest, then products are enzymatically treated, diluted and/or cleaned to remove unincorporated dNTPs and reduce salts which could interfere with analysis. During MS analysis of the PCR products, samples are ionized, then separated based on their mass-to-charge ratios. The ions are detected after laser desorption with a nitrogen laser. Mass spectra of PCR products are obtained by detecting positive ions of the nucleic acids. Differences in mass due to nucleotide base changes can be detected [21]. Mass spectrometry can detect low levels of sequence variations useful for detecting mosaicism, somatic changes in a normal background and heteroplasmy in mitochondrial DNA.
Example of Application of Mass Spectrometry
1.
Cystic fibrosis carrier testing
Multiplex Ligation-Dependent Probe Amplification (MLPA)
Deletions and duplications of single or multiple exons in specific genes are associated with many human diseases (reviewed in ref. 35). Although partial gene deletions or duplications account for less than 10 % of all disease-causing mutations for most hereditary conditions, some disorders can have deletion or duplication rates of 10–30 % or higher [36–44]. Multiplex Ligation-dependent Probe Amplification (MLPA, MRC Holland, Amsterdam, The Netherlands) is a semi-quantitative method used to detect abnormal copy numbers at an exon level resolution and has a high multiplexing capability [45]. The inclusion of MLPA in the clinical molecular laboratory can significantly increase the detection rate of many genetic disorders. Typically, MLPA kits contain a mixture of exon-specific probes targeted to the gene of interest and control probes that hybridize to other genomic areas.
In MLPA, DNA is denatured and incubated overnight with a mixture of probes that consist of two immediately adjacent oligonucleotides per target exon, each containing one of the PCR primer sequences (Fig. 2.5a). After hybridization, probes are ligated and the fragments are amplified by PCR using dye-tagged universal primers. Probes that are not ligated contain only one primer sequence and cannot be amplified to generate a signal. Amplification products that are typically between 130 and 480 bp in length, are separated by size using CE (Fig. 2.5a). The number of probe ligation products directly correlates to the number of target sequences in the sample. Deletions and duplications of the targeted regions are detected when the height ratios of the fluorescent peaks are lower or higher than the normal height ratio range of 0.7–1.4, respectively. An example of a large gene deletion where the deleted probes fall below the lower normal peak height ratio range of 0.7 is shown in Fig. 2.5b.
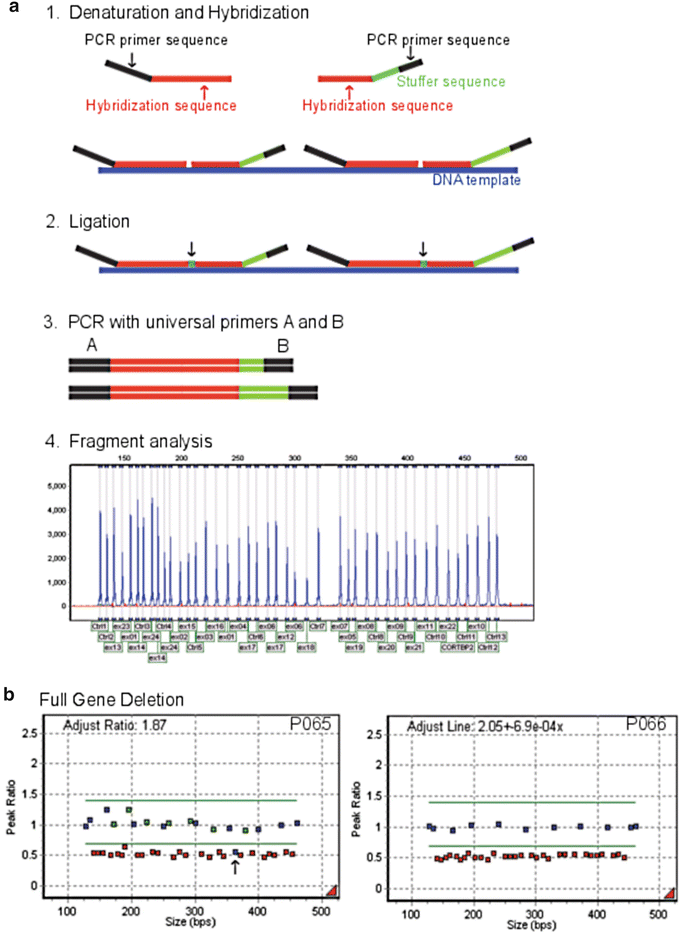
Figure 2.5
Multiplex Ligation-Dependent Probe Amplification (MLPA). In (a), the process of MLPA is shown. First, DNA template (blue) is denatured and then allowed to hybridize to exon specific probe hybridization sequences (red) which are targeted to the gene of interest. Adjacent probes are ligated together, and only ligated probes are amplified using universal primers A and B. Fragment analysis is used to separate the amplified fragments by size whereby the hybridization and stuffer sequences for each MLPA probe set determine the length of the amplified product. In (b), MLPA results of a large gene deletion are shown. Two MLPA kits (P065 and P066) were used to test for large FBN1 gene deletions and duplications that cause Marfan syndrome. An additional control probe (black arrow) located 301 Kb upstream from FBN1 exon 1 on the DUT (deoxyuridine triphosphatase) gene on chromosome 15q15-q21.1 also was deleted in this sample
Several variations on the traditional MLPA procedure have been developed. One example is reverse transcriptase MLPA (RT-MLPA) which can be used for mRNA profiling [46]. The only difference between traditional MLPA and RT-MLPA is that RT-MLPA begins with the reverse transcription of mRNA into cDNA before continuing with the typical MLPA reaction; the ligase enzyme cannot ligate probes which are bound to RNA. Methylation-Specific MLPA (MS-MLPA) is another variation that can be used to detect both copy number changes as well as the methylation status of the DNA target [47]. MS-MLPA is useful for imprinting disease testing [48–50] and the analysis of methylation aberrations in tumor samples [51, 52].
PCR Variations for Unknown Sequence Variants
Most of the techniques discussed above are used to screen for sequence variants (both mutations and polymorphisms) based on previous knowledge of the variant i.e., the sequence of the variant is either known or defined by previous scientific reports. In contrast, both research and clinical molecular pathology need methods to identify sequence variants without prior knowledge of their existence i.e., the sequence of the variant is unknown. Sequencing is the ultimate screening technique, but is costly and labor-intensive. The goal of the scanning techniques described below (denaturing gradient gel electrophoresis [DDGE], temperature gradient gel electrophoresis [TGGE], heteroduplex analysis [HA], single-strand conformation polymorphism [SSCP], denaturing high-performance liquid chromatography [DHPLC], protein truncation test [PTT], and variant screening by high-resolution melting [HRM] curve analysis) is to select DNA regions with possible variant sequences for follow up confirmation, thereby reducing costs relative to sequencing. Should an unknown variant be detected, for example by a shift in the mobility of the PCR product on a gel or capillary, the PCR product with altered mobility may be isolated and sequenced.
Denaturing Gradient Gel Electrophoresis and Temperature Gradient Gel Electrophoresis
Denaturing gradient gel electrophoresis (DGGE) [53, 54] and temperature gradient gel electrophoresis (TGGE) [55, 56] are similar methods for separating DNA fragments with similar lengths but different sequences and depends upon different mobilities within a linear gradient of increasingly denaturing conditions. In DGGE, the gradient is created with a mixture of urea and formamide, and in TGGE with a combination of water baths at different temperatures and a cooling plate under the gel. Both DGGE and TGGE exploit the markedly decreased mobility of partially melted dsDNA compared to either fully annealed dsDNA or ssDNA. Melting within a dsDNA fragment occurs within stretches of base pairs called melting domains. The point at which a domain begins to denature is referred to as the melting temperature (T m), whether melting was induced by temperature or denaturing chemicals. In general, GC-rich sequences are more resistant to denaturation because of the three hydrogen bonds holding a GC pair together, as opposed to the two hydrogen bonds in an AT base pair. During electrophoresis, once a dsDNA fragment reaches the point at which the melting domain with the lowest T m begins to denature, mobility of the fragment through the gel nearly ceases. Fragments that melt early in the gel can therefore be separated from those that melt later. Complete denaturation of the dsDNA can be prevented by adding a GC-rich region to the 5′ end of one of the primers (GC clamp), increasing the sensitivity for detection of sequence variants.
For DGGE and TGGE, the denaturing conditions and the time of electrophoresis are optimized such that non-variant sequences migrate to an intermediate position in the gel by the end of electrophoresis, allowing sequence variants creating either a higher or lower T m to be identified. The denaturing gradient may be perpendicular or parallel to the electric field. Perpendicular gradient gels covering a broad range of denaturing conditions are loaded with the non-variant sequence in all lanes to find the optimal, narrower denaturing gradient (chemical or temperature) for later use in parallel gradient gels. Parallel gradients are used to assess patient samples but also to optimize the time of electrophoresis by loading the non-variant sequence in different lanes at different times. Double-gradient DGGE adds a sieving gradient, for example, 6–12 % polyacrylamide, colinear with the denaturing gradient in the gel matrix, further improving band resolution.
Both DGGE and TGGE work best with DNA fragments less than 500 bp in length. When GC-clamped fragments are analyzed, the sensitivity of detecting a SNP is close to 99 %. Following electrophoresis, specific bands can be isolated from the gel and sequenced. DNA fragments with a high GC content are not easily analyzed by DGGE, since all fragments are more resistant to melting.
Examples of Applications of DGGE or TGGE
Heteroduplex Analysis
Heteroduplex formation results when non-variant and variant alleles are coamplified, denatured, and allowed to reanneal in a post-PCR annealing step [60]. Some of the strands pair with the complementary strand from the same allele and form homoduplexes. However, some strands pair with a strand from the other allele and form heteroduplexes. Because the heteroduplexes have mismatched base pairs between strands, they form a partially open dsDNA sequence that migrates more slowly during electrophoresis than the fully annealed homoduplexes.
Two types of heteroduplex structures can be formed [61]. When the mismatch consists of one or more single-base mutations, small open areas of dsDNA called “bubble-type” heteroduplexes are formed. When the mismatch is formed by insertions or deletions between the two alleles, a pronounced bending of the dsDNA is produced and referred to as a “bulge-type” heteroduplex. Bulge-type heteroduplexes markedly affect the mobility of the dsDNA, whereas bubble-type heteroduplexes may be difficult to detect electrophoretically. Detection of single base-pair bubble-type mismatches can be enhanced in two ways. Electrophoresis can be performed with mutation detection enhancement (MDE) gels, an altered form of polyacrylamide used for improved resolution. Alternatively, the post-PCR introduction of a known sequence with a short deletion to form a bulge-type heteroduplex enhances the separation of sequences with base-pair mismatches in a process known as universal heteroduplex generation.
Examples of Applications of Heteroduplex Analysis
1.
HIV subtyping
2.
CFTR gene mutation analysis for cystic fibrosis
3.




NF1 gene mutation analysis for neurofibromatosis type 1
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
