Gene
MSUD type
Locus
Size
Protein subunit
Exons
Founder mutations
BCKDHA
Ia
19q13
28 kb
E1a
9
p.T438N
BCKDHB
Ib
6q14
240 kb
E1b
11
p.R183P, p.G278S, p.Q372X
DBT
II
1p31
56 kb
E2
11
DLD
III (aka. LAD deficiency)
7q31
30 kb
E3
14
p.G229C
Clinical Utility of Testing
Molecular testing for MSUD confirms clinical and newborn screening results. Carrier screening for the Mennonite and Ashkenazi Jewish populations is available and allows for preconceptual and prenatal genetic counseling and for immediate treatment of an affected infant after birth.
Available Assays
Targeted mutation analysis of the common alleles via multiplex PCR is a cost-effective and quick method for the Mennonite and Ashkenazi Jewish populations, with a mutation detection rate of over 98 % for BCKDHA and BCKDHB, respectively [8]. For the general population, exon sequencing and deletion analysis of BCKDHA, BCKDHB, and DBT, and DLD are available.
Interpretation of Test Results
Mutations in BCKDHA, BCKDHB, and DBT do not have clear phenotype correlations, and treatment modality is essentially the same, regardless of the degree of biochemical phenotype. In a small study of 13 Ashkenazi Jewish families affected with LAD deficiency, homozygosity for the p.G229C allele appeared to be associated with a milder and later-onset disease, with no neurological manifestations [7].
Urea Cycle Disorders (Ornithine Transcarbamylase Deficiency)
Molecular Basis of the Disease
Defects in the urea cycle constitute a rare group of disorders resulting in the accumulation of urea precursors, mainly ammonium and glutamine. Ornithine transcarbamylase (OTC) deficiency, the most common inborn error of ureagenesis, is an X-linked disorder. Affected hemizygous males typically present in the neonatal period or later in childhood, with symptoms that include vomiting, lethargy, hypothermia, and apnea due to hyperammonemia, leading to coma or death. Recurrent episodes of metabolic crisis can result in ID. The only available treatment after an acute metabolic episode is liver transplantation, which should be performed as early as possible to prevent brain damage. In 15–20 % of carrier females, symptoms are evident. Symptomatic carrier females typically have a later onset but the disease can be fatal, presumably due to an unfavorable pattern of X-inactivation in the liver [9].
OTC is a homotrimeric mitochondrial matrix enzyme that catalyzes the synthesis of citrulline from ornithine and carbamyl phosphate, and is found almost exclusively in the liver and intestinal mucosa. Loss of OTC activity results in high plasma glutamine and ammonium, low plasma citrulline, and an excess of orotic acid in the urine, a combined metabolic profile that is diagnostic for OTC deficiency. However, a direct assay of OTC activity performed on tissue isolated from a liver biopsy specimen may be necessary to obtain unequivocal biochemical results.
The OTC gene is located on Xp21 and spans a region of 73 kb that contains ten exons and encodes a protein of 354 amino acids. The overall prevalence of the disease is estimated at 1 in 50,000 in the USA, with similar statistics reported in Japan. Mutations have been identified in all ten exons; however, disease-causing mutations are less frequent in exons 1 and 7, the least conserved exons, most likely reflecting their lesser relevance to the function of the enzyme [10].
Clinical Utility of Testing
Diagnosis of OTC deficiency by molecular testing is preferable to the more invasive liver biopsy that is necessary for the enzymatic test. Molecular screening for OTC mutations identifies approximately 80 % of mutations, while the remaining undetected mutations are expected to affect promoter function or splicing [11]. The OTC gene has an approximately 50:1 sperm-to-egg mutation rate ratio [9]. From 66–93 % of male probands inherit the mutation from their mothers, while only 20 % of manifesting females inherit the mutation, taking into account differences in new mutation rates between sperm and eggs, and lyonization effects for a female to manifest symptoms. Thus, a woman who has a son with OTC deficiency has a much higher chance of harboring the mutation than a woman who has an affected daughter. Due to the inheritability and the severity of the disease with the limited treatment available, molecular screening of at-risk couples for the purpose of prenatal testing may be beneficial. Additionally, mutation identification may be of prognostic value in OTC deficiency (see “Interpretation of Test Results” below).
Available Assays
Molecular testing using bidirectional sequencing of the entire coding region and intron-exon boundaries of the OTC gene is available in a number of clinical molecular laboratories. In addition, MLPA is used to detect large OTC coding region deletions/duplications. High-resolution aCGH can be used to detect exonic deletions/duplications or larger rearrangements that include the OTC gene as part of a contiguous gene deletion syndrome [12]. Approximately 300 mutations have been reported (http://www.cnmcresearch.org/OTC/). Most mutations (86 %) in the OTC gene are point mutations, with G to A transitions accounting for 34 % and C to T transitions accounting for 21 % of the total. Approximately one-third of all point mutations are at CpG dinucleotides, and 15 % are at splice junctions. Although the CpG sites are recurrent mutation sites, none accounts for more than 4 % of the total single-base substitutions.
Interpretation of Test Results
In general, the genotypic spectrum correlates with the severity of the phenotype, and mutations that result in complete loss of function or amino acid changes near the active site of the protein result in neonatal onset of disease. In contrast, amino acid changes that are not close to the active site and result in protein with residual enzymatic activity are associated with later onset and a milder disease course [12].
Lysosomal Storage Disorders
Lysosomal storage disorders are a group of diverse inherited metabolic diseases that result from the disruption of the lysosomal system and catabolism of macromolecules (for review, see Ref. 13). Mutations in genes encoding hydrolyzing enzymes, activator proteins, lysosomal membrane proteins, or proteins involved in the posttranslational modification or transport of lysosomal proteins can cause such storage disorders. More than 40 lysosomal storage disorders are known, and they have a collective incidence of approximately 1 in 5,000–8,000 live births in the USA. Most of the genes responsible for lysosomal storage disorders have been cloned, permitting molecular testing once a diagnosis is established by biochemical analyses. This information is valuable for genotype-phenotype correlation, selection of therapy, and genetic counseling. In this section, two lysosomal storage disorders are discussed: Tay-Sachs disease (TSD), which serves as a model for population screening, and Gaucher disease (GD), for which much effort has been concentrated on genotype-phenotype correlations.
Tay-Sachs Disease
Molecular Basis of the Disease
TSD is a neurodegenerative disorder resulting from deficiency of the lysosomal enzyme hexosaminidase A (HEX A), resulting in accumulation of the cell membrane glycolipid GM2 ganglioside within lysosomes (for review, see Refs. 14, 15). The clinical course of TSD is characterized by normal development for the first few months of life followed by progressive loss of motor skills, macrocephaly, seizures, blindness, and death usually before 4 years of age. Infantile TSD always is fatal and there is no effective treatment. Later-onset forms of TSD have slower disease progression. TSD is an autosomal recessive disease and has a carrier frequency of approximately 1 in 30 among Ashkenazi Jewish individuals and 1 in 250–300 in most other populations. Genetically isolated populations such as the French Canadians of Quebec, Cajuns from Louisiana, and the Amish in Pennsylvania also have carrier frequencies similar to the Ashkenazi Jews. The first carrier screening programs began in 1970 and used the measurement of HEX A activity in serum, leukocytes, or tears. When the HEXA gene encoding HEX A was cloned in 1987, disease-associated mutations were identified. Current testing for TSD utilizes both biochemical and molecular testing by various methods.
Clinical Utility of Testing
Carrier screening for TSD, which began in the 1970s and was later endorsed by the American College of Obstetricians and Gynecologists (ACOG) and the ACMG, has been a model for population screening programs. As a result, there has been a 90 % reduction in the incidence of TSD in the North American Ashkenazi Jewish population, such that the incidence of TSD is now three- to fourfold higher in non-Jews by comparison.
Available Assays
Clinical laboratories use several strategies to incorporate mutation testing into their screening programs. Some laboratories initially screen by enzyme analysis followed by molecular testing for individuals with a result in the carrier or inconclusive ranges. Other laboratories use molecular testing alone for selected populations. Molecular tests are performed using a variety of methods, including PCR amplification followed by allele-specific primer extension analysis, allele-specific oligonucleotide (ASO) hybridization or restriction enzyme digestion, allele-specific amplification, TaqMan probe technologies, or ligation chain reaction amplification. In Ashkenazi Jewish individuals, two common mutations in HEXA are associated with infantile TSD and one associated with an adult-onset form of the disease. A four-base pair insertion (c.1274_1277dupTATC) in exon 11 accounts for approximately 80 % of mutant alleles in the Ashkenazi Jewish population, and a splice defect in intron 12 (c.1421+1G>A; IVS12) accounts for another 15 %. A missense mutation, p.G269S, leads to an adult-onset form of TSD and accounts for approximately 2 % of carriers.
Interpretation of Test Results
A pseudodeficiency allele, p.R247W, is present in approximately 2 % of Ashkenazi Jewish individuals who are carriers by the enzymatic assay. The p.R247W variant decreases the activity of HEX A for the artificial substrate used in the laboratory but does not cause TSD since it does not affect HEX A activity for its natural substrate, GM2 ganglioside. About 36 % of non-Jewish individuals who are carriers by enzyme analysis have a pseudodeficiency allele (32 % p.R247W and 4 % p.R249W). In addition, screening for the three common Ashkenazi Jewish mutations and an additional mutation (c.1073+1G>A; IVS9) will identify approximately 95 % of Ashkenazi Jewish carriers, but only 40–50 % of disease-causing alleles in the non-Ashkenazi Jewish populations. Other populations who are at high risk, such as the French Canadians, Cajuns, and Pennsylvania Dutch, have their own common alleles. Therefore, the mutations included for population screening must target the specific mutations of the ethnic background of the individuals being tested.
Gaucher Disease
Molecular Basis of the Disease
GD is another prevalent autosomal recessive lysosomal storage disorder that is found with higher incidence in the Ashkenazi Jewish population. The carrier frequency is 1 in 15 in the Ashkenazi Jewish population and 1 in 100 in other populations [16] (for comprehensive review on GD, see Ref. 17). A defect in the enzyme glucocerebrosidase leads to the accumulation of glucocerebrosides in lysosomal compartments in macrophage/monocyte-derived cells, particularly in the liver, bone marrow, spleen, and lung. Several forms of GD exist. Type 1 GD has a wide range of clinical presentations, from asymptomatic to symptoms including bone disease, hepatosplenomegaly, anemia, and thrombocytopenia, but without primary central nervous system involvement. Types 2 and 3 have primary central nervous system involvement that varies by age of onset and rate of disease progression. Type 2 GD patients usually have an earlier age of onset than type 3 patients, with acute disease progression and death by approximately 2 years of age. Type 3 patients have onset in early childhood to adolescence and survive into their first to fourth decade of life. A perinatal-lethal form of GD also can occur, as well as a cardiovascular form characterized by aortic and mitral valve calcification, ophthalmologic abnormalities, and hydrocephalus.
The glucocerebrosidase gene (GBA) and its transcribed pseudogene (ψGBA) are located on chromosome 1q21. Approximately 200 mutations causing GD have been identified. Many of the mutations are most likely due to gene conversion events with the pseudogene.
Clinical Utility of Testing
The demonstration of deficient glucocerebrosidase activity in leukocytes establishes a diagnosis of GD but is unreliable for carrier detection. Therefore, molecular testing is useful for carrier identification, prenatal testing, and genetic counseling.
Available Assays
Four mutations (p.N409S, c.115+1G>A, c.84dupG, p.L483P) are responsible for approximately 95 % of disease-causing alleles in Ashkenazi Jewish individuals and 50 % of disease-causing alleles in non-Ashkenazi Jewish individuals [16]. Most clinical molecular laboratories performing GD molecular testing assess at least these four mutations. Several factors must be considered when designing molecular testing for GD. Primers must be selected that avoid amplification of the pseudogene which is located 16 kb downstream and is approximately 96 % identical to the functional gene. Recombinant alleles, which are thought to result from unequal crossovers between exons 9 and 10 of the functional gene and pseudogene, contain two or more point mutations, including p.L483P. If p.L483P alone is tested, misdesignation of the genotype may occur. This may be important, as the recombinant allele is typically associated with a more severe genotype. Mistyping also is possible when a 55 base pair (bp) deletion in exon 9 (c.1263_1317del), is present in combination with the common p.N409S allele. Homozygosity of p.N409S would be observed even though the true genotype is p.N409S/55 bp deletion. Therefore, the 55 bp deletion should be analyzed in patients who are found to be homozygous for p.N409S.
Interpretation of Test Results
Genotype-phenotype correlations have been widely investigated in GD [18]. While overlaps occur, some generalizations can be made. The presence of a p.N409S allele is predictive of type 1 disease. Individuals with p.L483P in the presence of a null allele will usually have type 2 GD, while homozygosity for p.L483P typically results in type 3 GD. Homozygosity for the p.D448H allele has been associated with the rarer cardiovascular form of GD.
Disorders of Carbohydrate Metabolism
Galactosemia, fructosemia, and the glycogen storage diseases are discussed in this section, which addresses disorders of carbohydrate metabolism.
Galactosemia
Molecular Basis of the Disease
Galactosemia is an autosomal recessive disorder caused by deficient or absent activity of one of three enzymes involved in the metabolic pathway to convert galactose to glucose: galactokinase (GALK), galactose-1-phosphate uridyl transferase (GALT), and UDP-galactose 4′-epimerase (GALE). Classic galactosemia is due to a severe reduction or absence of the GALT enzyme, and has an incidence of 1 in 40,000–60,000 in European newborns (for review, see Ref. 19).
The symptoms of classic galactosemia in neonates include poor feeding, vomiting, failure to thrive, lethargy, jaundice, occasionally diarrhea, and E. coli sepsis. The symptoms in an affected newborn can be obviated if a lactose-free diet is initiated within the first 2 weeks of life. Newborn screening for galactosemia is included in all 50 states. Newborns with a positive screen are followed up with immediate dietary treatment and confirmatory biochemical analysis.
The GALT gene is located at 9q13, is about 4 kb in length, and consists of 11 exons. More than 150 mutations in the GALT gene have been reported, most of which are private mutations [20]. The p.Q188R mutation is the most frequent mutation associated with classic galactosemia in many populations, and accounts for 64 % of disease alleles in Europeans, 60–70 % in Americans, and 50–58 % in Mexican Hispanics [20, 21]. Ethnic-specific mutations include p.K285N, p.S135L, c.253-2A>G, and a 5 kb deletion in Caucasian, African-American, Hispanic, and Jewish patients, respectively.
Clinical Utility of Testing
Molecular testing is used for confirmation of diagnosis, carrier detection, prenatal diagnosis, prognosis, and genetic counseling. Because the detection rate of molecular testing is less than 100 % and biochemical testing is highly accurate, mutation detection is carried out in parallel with biochemical analysis.
Prenatal diagnosis for galactosemia can provide the opportunity for immediate dietary restriction of the newborn. Although galactosemia is considered “treatable,” symptoms such as ID, growth delays, speech dyspraxia, abnormal motor function, and premature ovarian failure in women may still occur even with early intervention and lifetime dietary restrictions. These long-term phenotypes often are associated with specific mutations; for example, p.Q188R can cause premature ovarian failure and speech dyspraxia. Genetic counseling is very important for parents of affected fetuses before a choice regarding pregnancy outcome is made.
Available Assays
Initial molecular testing for the diagnosis of galactosemia focuses on the most prevalent mutations or the p.N314D mutation associated with Duarte galactosemia (Duarte-2) by quick and cost-effective methods, such as multiplex PCR followed by restriction enzyme digestion [22]. If only one mutation or no mutations are found, screening of all 11 exons and exon-intron boundaries by DNA sequencing can be performed [22]. A detection rate of 96 % can be achieved by a combination of testing for prevalent mutations and DNA sequencing [23]. Gross deletions and duplications are detected by aCGH or MLPA.
Interpretation of Test Results
Genotype-phenotype associations have been established for some mutations [20]. For example, p.Q188R, p.K285N, and p.L196P alleles have undetectable GALT activity and are associated with severe phenotypes, whereas p.S135L and p.T138M are less severe and are usually associated with a good prognosis. In addition to the classic form of galactosemia, which has less than 5 % of the normal GALT activity, the Duarte-2 variant associated with the p.N314D variant has 50 % of the normal GALT activity. The p.N314D allele is in linkage disequilibrium with four polymorphisms, c.329-27G>C, c.378-24G>A, c.507+62G>A, and c.-119_-116delGTCA. The symptoms of Duarte-2 patients are mild compared to the classic type, but Duarte-2 still results in long-term phenotypes such as ID and growth delays, speech dyspraxia, abnormal motor function, and premature ovarian failure in women.
Hereditary Fructose Intolerance
Molecular Basis of the Disease
Hereditary fructose intolerance (HFI) is an autosomal recessive disorder caused by deficient or absent activity of aldolase B. The aldolases are a group of tetrameric enzymes that are highly conserved and involved in the cleavage of fructose-1,6-biphosphate. Aldolase B also cleaves fructose-1-phosphate, and thus is a key player in fructose metabolism, as well as in gluconeogenesis and glycogenolysis. Aldolase A is found predominantly in muscles, whereas aldolase B is found predominantly in the liver, kidney, and intestine, and aldolase C is predominantly in the brain. Severe reduction or absence of only the aldolase B enzyme causes HFI, and has an incidence of 1 in 20,000–30,000 in European newborns (for review, see Ref. 24).
Typically in HFI, symptoms do not arise until an infant is exposed to fructose, sucrose, or sorbitol-containing foods, usually at the weaning of breast milk and introduction of table foods. Acute ingestion of the offending sugar can result in a variable presentation of abdominal pain, vomiting, progressive liver dysfunction, hypoglycemia, uric and lactic acidosis, and renal tubulopathy, depending on the dose of the sugar ingested, and timing and length of exposure. Chronic, low-dose ingestion of fructose-containing foods can cause a subacute picture of hepatomegaly and failure to thrive. Affected individuals typically have an aversion to sweets. The acute and chronic symptoms are, for the most part, reversible, if a fructose-free diet is initiated. Once on treatment, the prognosis is good with a normal life expectancy.
The ALDOB gene is located at 9q22.3, is about 14 kb in length, and consists of nine exons, eight of which are translated. More than 40 mutations have been identified (http://www.bu.edu/aldolase/HFI/hfidb/hfidb.html). The most common allele, p.A149P, accounts for 50–65 % of all mutations in the European population, and together with two other common mutations, p.A174D and p.N334K, account for more than 80 % of mutant alleles in the European population [25]. Most of the missense mutations are located in exons 5 and 9. Studies show that the p.A149P decreases the thermal stability of the enzyme. Figure 11.1 illustrates the locations of the most common variants in the aldolase B protein structure.
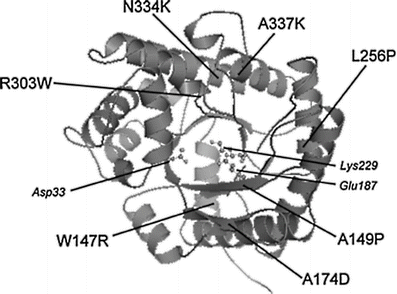
Figure 11.1
Location of some naturally occurring aldolase B mutations, shown on the crystal structure of human aldolase B (based on Protein Data Bank [PDB] structure file 1QO5, chain A) [26]. Only one monomer is shown for clarity. Active site residues are shown in a ball and stick representation and labeled in italics. Reprinted from Bouteldja N, Timson DJ. The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis 2010;33(2):105–12 [24], by permission of Springer
Clinical Utility of Testing
Molecular testing is used for confirmation of diagnosis, carrier detection, prenatal diagnosis, and genetic counseling. Enzymatic testing of aldolase B activity can only be performed from liver, kidney, or intestinal tissue. Thus, molecular testing is now the preferred method. Prenatal diagnosis for HFI can provide the opportunity for early dietary restriction and prevention of acute symptoms and liver disease.
Available Assays
Targeted mutation analysis of the common alleles using multiplex PCR is a cost effective and rapid method for the European population [27]. If only one mutation or no mutations are found, DNA sequencing of the entire coding region and exon-intron boundaries should be performed. For those not of European descent, DNA sequencing should be performed as a first-line method.
Interpretation of Test Results
Genotype-phenotype correlations are not clear. HPI patients with the same genotype have varied presentations and reactions to ingestion of fructose.
Glycogen Storage Diseases
Molecular Basis of the Disease
Glycogen storage diseases (GSD) are a group of heterogeneous genetic disorders characterized by the accumulation of glycogen in tissues and have an overall incidence of 1 in 20,000–25,000 live births [28]. Fourteen types of GSD that vary significantly in clinical phenotypes, age of onset, and affected organs have been identified (www.omim.org). They are caused by defects in one of 18 genes in glycogen metabolism. A summary of the 14 GSD types is presented in Table 11.2. The most common and severe types, GSD I, II, III, and IV, are discussed below.
Table 11.2
Glycogen storage disease types and subgroups
Disorder(s) | Defective enzymes | Gene symbol | Gene location | Inheritance pattern |
|---|---|---|---|---|
GSD I (von Gierke disease) | GSD1a: Glucose-6-phosphatase, catalytic | G6PC | 17q21.31 | AR
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|