Figure 32.1
Approximate prevalence of identified “driver mutations” in unselected pulmonary adenocarcinoma. While many of these alterations are typically seen in isolation, others, such as PIK3CA, are often seen concurrently with other gene mutations. The spectrum of changes is different from that seen in squamous cell carcinoma of lung and varies by smoking status. Asterisk indicates a rearrangement or amplification; dagger indicates mutation [41] Ding L et al. 2008
Clinical Utility of Testing
Molecular testing significantly drives clinical therapeutic decision making for patients with NSCLC. Three commonly performed molecular tests, EGFR mutation testing, KRAS mutation testing, and ALK rearrangement testing usually by fluorescence in situ hybridization (FISH), illustrate three different scenarios for the clinical utility of molecular testing in NSCLC.
EGFR TKIs, including erlotinib and gefitinib, gained regulatory approval in unselected populations of NSCLC patients in the USA and Europe. The response rate in early studies of unselected lung cancer patients was only 10–15 % but was of marked degree in those patients who did respond. Later, activating EGFR mutations were identified to confer sensitivity to EGFR TKIs and accounted for the subset of tumors with response to therapy in early studies [9, 37] and that such therapy results in significantly higher overall response rates and progression free survival compared to standard chemotherapy. For patients without “sensitizing” EGFR mutations, chemotherapy without TKIs was clearly superior [37–39]. Thus, despite the broad indication for these drugs, the use of EGFR TKIs has dramatically shifted to their preferential early use in patients with proven “sensitizing” EGFR mutations. Patients without a “sensitizing” EGFR mutation also can benefit from the use of EGFR TKI in maintenance or later lines of therapy, but the benefit is modest [40, 41].
The oral kinase inhibitor, crizotinib, first entered clinical trials as a MET inhibitor for patients with MET gene amplification or activating mutations. After the discovery of ALK gene fusions in approximately 5 % of NSCLC patients, the trial was amended to include patients who demonstrated evidence of an ALK gene rearrangement by FISH. Following phase I and II data with overall response rates of 55–63 %, crizotinib gained provisional FDA approval for this highly selected group of patients [34, 42]. Unlike EGFR TKIs, which are approved for all patients with pulmonary adenocarcinoma, crizotinib is only approved for the treatment of patients with lung cancer carrying an ALK rearrangement, although its utility in other molecular subtypes, such as ROS1 rearrangement-positive tumors, is under investigation [44, 45].
KRAS activating mutations occur in approximately 25 % of patients with NSCLC. Unfortunately, no KRAS-targeted therapies are currently FDA approved. The presence of a KRAS mutation, however, is still informative. The overlap of KRAS mutations with either sensitizing EGFR mutations, ALK gene rearrangements, or other oncogenic abnormalities is infrequent [46]. The presence of a KRAS mutation thus augments the negative predictive value of a negative EGFR mutation test or a negative ALK FISH test. Likewise, the presence of a KRAS mutation predicts a lack of tumor response from an EGFR TKI, although it does not necessarily predict a complete lack of benefit from an EGFR TKI [40, 41]. The detection of KRAS mutations can identify patients for participation in clinical trials. Currently, inhibition of MEK and PI3K pathways, both of which are downstream of KRAS, is under investigation in lung and other cancers. Thus, directing patients with a KRAS mutation-positive lung cancer to these clinical trials may enrich for patients who are likely to benefit from these strategies. Conversely, the presence of a KRAS mutation may provide a useful negative selection criterion for clinical trials that are targeting pathways upstream of KRAS [47].
In summary, molecular testing can provide guidance for prioritizing therapies to those patients most likely to receive the greatest benefit (EGFR mutations). Alternatively, molecular testing can identify patients with proven benefit from a targeted therapy (ALK gene rearrangements). Finally, molecular testing can provide clinicians with a rationale to guide certain patients toward clinical trials of targeted therapeutics (KRAS mutations). These selected examples of targets are by no means the only ones of interest in NSCLC, in which a growing number of molecularly targeted agents are under investigation in association with specific molecular alterations. This includes mutations in BRAF, HER2, PIK3CA, and rearrangements in RET, among others [47].
Available Assays
Specimen Type, Quality, and Tumor Content
A discussion of the practical elements of molecular testing in NSCLC necessitates an understanding of the variability of specimen types likely to be encountered. As the majority of NSCLC patients present with advanced disease, they are not surgical candidates, and the diagnostic tumor sample is often the only tissue available for testing. This has driven the need to undertake evaluation of small specimens obtained from diagnostic biopsies, fine needle aspirations (FNA), and body cavity fluids. Cytopathology specimens are suitable for molecular testing, including cell block preparations as well as smeared and imprinted slides [49–55]. For small specimens, the quality of the specimen is often more important than the quantity [52, 56]. Individual laboratories are responsible for validation of the adequacy of all specimen types for each test.
Of paramount importance for any specimen type is the evaluation of the specimen by a pathologist with experience in the assessment of pathology specimens for molecular analysis. The tumor content of the specimen should be assessed for the presence of adequate non-necrotic tumor cells, as well as an estimation of the percent of tumor cells relative to all nucleated cells, which meets the criteria for acceptability for testing set by the clinical molecular laboratory [53]. Features suggesting that the specimen may not be of optimal quality include extensive necrosis, paucity of tumor cells, low abundance of tumor cells compared to other non-tumor cells in the specimen, and exposure to reagents which negatively impact molecular testing (e.g. decalcification) [52, 57, 58].
Some specimens are not sufficiently tumor rich to be utilized without tumor enrichment methods. Tumor enrichment methods include manual macrodissection, manual microdissection, and laser capture microdissection (LCM) [59]. Manual macrodissection is when a region of interest is scraped from a slide or carved from a paraffin block without microscope assistance. Manual microdissection is performed with a dissecting or light microscope, often using a scalpel, needle tip, or hollow bore pipette to preferentially isolate tumor cells and using a microscopically marked guide slide. LCM utilizes computer-aided imaging to select specific cells for capture from specially coated slides. However, the cost of equipment and specialized consumables, combined with a labor intensive process, makes LCM the costliest approach to tumor enrichment, and assays typically do not require this degree of precision. However, in some circumstances, the use of LCM may increase the ability to utilize low-sensitivity assays for some specimen types [60–62].
Common Driver Mutations Assays
Evaluation of mutation status in genes such as EGFR and KRAS can be accomplished using a variety of platforms. Historically, Sanger sequencing has been the “gold standard” for EGFR and KRAS mutation detection. Because of the comparatively low analytic sensitivity of Sanger sequencing, numerous other platforms have been developed with higher analytic sensitivity but typically assess only specific known mutations (Fig. 32.2) [56, 63]. The compromise for these mutational assays is the loss of detection of rare alterations. However, the clinical relevance of rare mutations is often not well established, and thus, many clinical laboratories find that the ease of use of a targeted assay, combined with improved analytic sensitivity (thus reducing the degree to which tumor enrichment must be achieved), is an acceptable trade-off. Owing to the rapid evolution of the recommendations for which specific genes and alleles should be tested, specific recommendations are not elaborated herein, and recent guidelines should be consulted [64]. The more common methods for cancer gene mutation testing are described here.
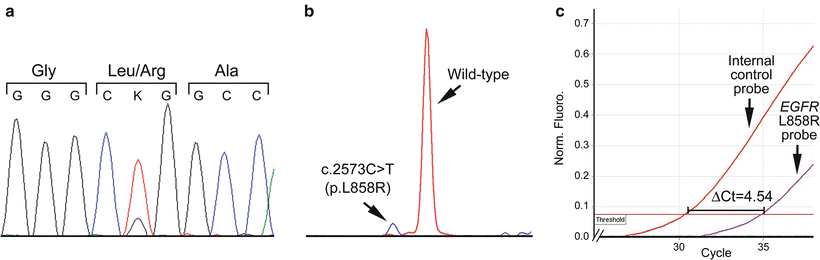
Figure 32.2
Different analytic sensitivities of mutation detection methods. (a) Sanger sequencing showing an EGFR c.2573T > G (p.L858R) mutation at 12.5 % allelic burden. (b) Single nucleotide base extension assay (SNaPshot®) showing the same mutation detectable at 6 % allelic burden. (c) Real-time PCR assay demonstrating the same mutation detectable at 1 % allelic burden
Sanger Sequencing
The primary advantage of Sanger sequencing for somatic mutation detection in NSCLC is the comprehensive assessment of the sequenced gene regions. The importance of comprehensive gene mutation assessment differs for different genes. Some of these assessments are influenced by the degree to which a specific target gene has been evaluated. For example, extensive evaluation of mutations in EGFR in NSCLC has demonstrated that a small number of mutations comprise the majority of changes. Thus, comprehensive assessment may only seldom identify a rare mutation not detected using targeted assays. In contrast, evaluation of mutations in DDR2 in squamous cell carcinoma has been undertaken to a lesser degree in recent years, and the data to date suggest a wide distribution of mutations, indicating that the advantages of Sanger sequencing are indeed relevant.
Modifications to Sanger Sequencing
A number of modifications to Sanger sequencing, including co-amplification at lower denaturation temperature-PCR (COLD-PCR) [65, 66] and improve and complete enrichment COLD-PCR (ICE-COLD-PCR) [67], leverage the advantages of Sanger sequencing, i.e., comprehensive assessment, while achieving improved analytic sensitivity. These methods typically utilize alterations in cycling parameters to achieve imbalanced amplification favoring mutated alleles, thus increasing analytic sensitivity, followed by Sanger sequencing.
Restriction Fragment Length Polymorphism and PCR Fragment Sizing
A common method for EGFR mutation detection is combined PCR fragment sizing and restriction fragment length polymorphism (RFLP) for exon 19 deletions and L858R (c.2573C > T), respectively [68]. This approach is straightforward and has an analytic sensitivity of approximately 5 % mutant allele detection and excellent ease of use. The major disadvantage to this approach is the limitation in mutations detected, specifically that a number of mutations in EGFR which are not common, but similarly are not exceptionally rare, are not detected by this approach. Recent guidelines suggest that all mutations which are seen with a frequency of at least 1 % of EGFR-mutated lung adenocarcinomas, should be included in routine screening, which would not be accomplished by this approach [64].
Real-Time PCR
Multiple (non-FDA approved) commercial real-time PCR test kits are available for the detection of specific mutations in some genes, including EGFR, KRAS, and BRAF. Many of these kits indicate an analytic sensitivity of approximately 1 % mutant allele detection. A major advantage of real-time PCR is the high analytic sensitivity, which therefore reduces the likelihood of a false-negative result based on insufficient tumor content in the specimen [63]. In mid-2013, two real-time PCR-based assays (therascreen EGFR RGQ PCR Kit [Qiagen, Manchester, the United Kingdom] and cobas® EGFR Mutation Test [Roche, Pleasanton, CA]) were approved by the FDA for the evaluation of common EGFR mutations.
PCR Screening Methods
High-resolution melting curve analysis and denaturing high-performance liquid chromatography allow for rapid assessment of whether an alteration is present in a targeted region but do not identify the specific gene variation [69, 70]. The major advantage of mutation screening approaches is a rapid initial assessment of the status of a target region. However, precisely defining the alteration requires a follow-up test method to confirm and delineate the finding.
Pyrosequencing
Pyrosequencing has greater analytic sensitivity compared to Sanger sequencing and, rather than a purely targeted approach, does allow for the evaluation of short stretches of sequence. Pyrosequencing can be used for the detection of most single base substitutions and, although more challenging to interpret, can also be utilized for deletions and insertions (such as are commonly seen in EGFR) [71, 72]. The major advantages of pyrosequencing include better analytic sensitivity, ability to design custom nucleotide dispensations to query specific changes, and comprehensive evaluation over short stretches of sequence.
Ultrahigh Sensitivity Methods
Specific alterations may indicate the need for ultrasensitive methods for detection. One example of this is EGFR T790M (c.2369C > T), which is known to exist at varying percentages within the tumor cell population, due to both variable degrees of EGFR amplification and subclonality of the alteration. Multiple methods to achieve analytic sensitivities below 1 % allelic burden include locked nucleic acid assays and PCR assays utilizing restriction endonucleases to cleave wild-type sequences and beads, emulsion, amplification, and magnetics (BEAMing) [73–77]. The implementation of these approaches is dependent upon the clinical need for enhanced sensitivity in mutant allele detection, for example, in approaches which evaluate circulating tumor DNA, which are under evaluation [76].
Multiplex Methods
Increasingly, test methods which allow for the simultaneous detection of many mutations in a number of genes are used by clinical molecular laboratories. These include multiplexed single nucleotide base extension assays and mass spectrometry-based assays [78–80]. These assays rely on the development of multiplex PCR amplification followed by interrogation by multiple probes with spacer sequences and base extension reactions to allow for discrimination based on size or molecular weight. In addition, next generation sequencing (NGS) can query numerous entire genes, and the implementation of this approach into the clinical molecular laboratory is undergoing rapid adoption with the evaluation of appropriate metrics for quality assurance [81, 82]. Importantly, depending on methodology utilized for NGS, an assay may be able to identify selected gene rearrangements and copy number alterations.
Gene Rearrangements Assays
FISH Assays
The EML4-ALK fusion is generated by a paracentric chromosomal inversion in the short arm of chromosome 2, between bands p21 and p23.2. These two genes originally are separated by approximately 12.8 MB. The other described ALK fusions in lung cancer, KIF5B–ALK [34] and TFG–ALK [33], are caused by chromosomal translocations t(2;10)(p23.2;p11.22) and t(2;3)(p23.2;q12.1), respectively.
Numerous technical platforms are available for molecular diagnosis of ALK rearrangements in lung cancer, but the most commonly used for clinical testing is FISH. The standard FISH assay uses a dual-color break-apart probe set encompassing the 3′ and the 5′ sequences immediately adjacent to the breakpoint area in ALK (intron 19), labeled in orange and in green fluorophores, respectively (Fig. 32.3) [83].
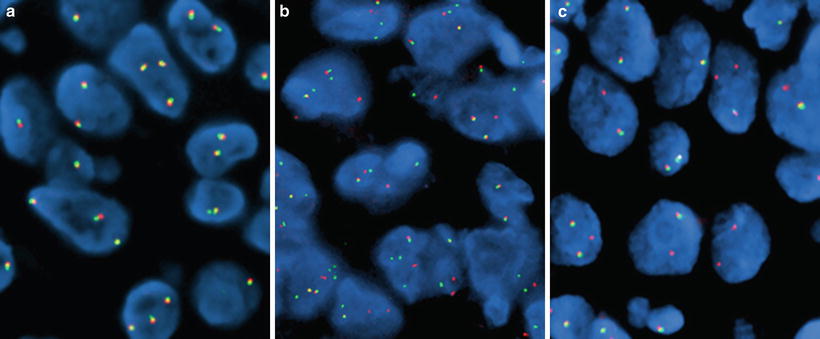
Figure 32.3
Lung adenocarcinoma specimens hybridized with the Vysis ALK Break Apart FISH Probe (Abbott Laboratories, Abbot Park, IL). (a) ALK-negative pattern (only native orange/green fused fluorescent signals), (b) ALK-positive pattern (split orange-green signals), and (c) ALK-positive pattern (single orange signals)
The ALK BA FISH probe was used in the initial clinical trials for crizotinib [35, 84] and is approved by the FDA as a companion diagnostic test. The ALK BA FISH assay is DNA based and is thus robust and resistant to technical artifacts. Other advantages include being highly suitable for detection in FFPE tissue sections, detecting all variants of EML4-ALK, and detection of translocations with and of the reported or uncharacterized partner genes. On the other hand, the FISH platform requires specialized resources (fluorescence microscope, light-protected laboratory space) and highly trained personnel.
Two other receptor kinase genes were identified as activated in lung cancers by fusions, ROS1 [33] and RET [85]. Integrated molecular and histopathologic screening systems have been successful in detecting a large number of fusion partners in lung cancer for these two genes, and preclinical studies have confirmed their tumorigenic potential [34]. Additionally, targeted therapy may have a role for these molecular subsets, which are predominantly identified using FISH assays [44, 45, 86].
Reverse-Transcription PCR
Reverse-transcription PCR (RT-PCR) can be used to detect and identify gene rearrangements. Messenger RNA (mRNA) is isolated from tumor tissue and reverse transcribed to complementary DNA (cDNA). Amplification with polymerase is performed using primers that bind sequences within the 5′ gene fusion partner (e.g., EML4) and the 3′ end of the fusion gene encoding the kinase domain of the oncogene (e.g., ALK). The presence of a gene fusion can thus be detected and identified by size discrimination assays and sequencing of the amplification product. To date, over ten EML4-ALK variants have been reported, involving eight different EML4 exons and exon 20 of ALK [31]. Although methods exist to identify unknown gene fusion partners, such as inverse PCR and 5′ rapid amplification of cDNA ends (RACE), high-throughput targeted assays will typically only allow known fusion partners and variants to be detected [32]. Specific primers are designed for each EML4 variant, which can be multiplexed for PCR amplification, as long as the amplicons are of different sizes and/or labeled with different fluorophores. Amplicon size must be kept relatively small (up to 150–200 base pairs) to allow the use of FFPE tissue, since frozen tissue is not routinely available. The implementation of these assays in the clinical molecular laboratory requires either follow up on testing for negative findings (i.e., extensive analysis of RNA quality), separate amplifications for each variant, or design of assays to minimize amplicon size [32].
Other Methods
Immunohistochemistry (IHC) using antibodies specific for the C-terminal region of the ALK protein is a surrogate method to detect the presence of an ALK gene rearrangement in NSCLC [87, 88]. The most commonly used antibodies are ALK1 (DAKO, Carpinteria, CA), 5A4 (Novocastra, Newcastle, the United Kingdom, and Abcam, Cambridge, MA), and D5F3 (Cell Signaling Technology, Danvers, MA). IHC is a favored test method due to rapid turnaround time and low cost. However, for ALK IHC, the methods are not standardized, and method variations include antibody dilutions, detection systems with or without signal amplification, scoring criteria (qualitative, semiquantitative, and image-based), and cutoffs for classifying positive specimens, limiting the utility of IHC in the clinical molecular laboratory. However, extensive study is underway to define the concordance and reproducibility of this methodology with the expectation that following large-scale evaluations, IHC may become a dominant approach [89, 90, 91].
Quantitative real-time PCR (qPCR) techniques, using primers that amplify a portion of the 3′ end of the ALK gene (encoding the kinase domain), detect higher ALK mRNA expression in comparison with a housekeeping gene [92]. Both IHC and qPCR rely on the observation that ALK is not typically expressed in normal lung tissue or in NSCLC without an ALK gene rearrangement. Both techniques thus infer the presence of an ALK gene rearrangement but could also detect aberrant expression of native, full-length ALK.
Gene Amplification Assays
Oncogene activation is also achieved through amplification of gene copies. Mechanisms for gene amplification include the extrachromosomal double minutes and the intrachromosomal homogenously staining regions, both of which are easily detected by FISH assays.
In NSCLC, numerous genes can be amplified and typically confers a poor prognosis. Amplification of the EGFR gene is associated with response to EGFR inhibitors [93], although, at least in patients of Asian ethnicity, this association may largely reflect the amplification of activated mutant alleles [94] and the degree of this association is relatively weak compared to the evaluation of EGFR mutation status. Amplification of the MET gene has been associated with poor overall survival [95] and with acquired resistance to EGFR TK inhibitors [96], which is thought to arise from positive selection of low-level clones carrying the alteration [97]. MET amplification evaluation may also have relevance for the selection of targeted therapy as studies evaluating this analyte with respect to inhibitors of hepatocyte growth factor receptor (HGFR, the protein product of the MET gene) are ongoing [48]. A number of other genes encoding growth factors, growth factor receptors, and transcription factors can be amplified in lung cancer, such as HER2, FGFR1–FGFR2, SOX2, and PIK3CA. The availability of novel therapeutic agents that specifically inhibit these proteins has created an increased interest in testing for these molecular markers in order to select patients who are more likely to be sensitive (or resistant) to the targeted therapies [48].
Interpretation of Results
Driver Mutation Detection Assays
One of the key issues in the interpretation of somatic mutation testing is correlation between the preanalytic specimen features, preanalytic processing (especially tumor enrichment), testing method, and test results. This is especially relevant for negative results, which should be interpreted in the context of the above-listed features. Given that assessments of tumor cellularity are highly subjective and only a single specimen may be available for testing, a laboratory may elect to test a borderline specimen with a low percentage of tumor cells, because a positive result would be informative, even though a negative result may be inconclusive.
With gene sequencing assays, the interpretation of rare mutations can be challenging. For many gene targets, mutational hotspots have been identified, and a finding of a mutation outside of one of these known regions must be carefully interpreted for clinical significance. Since FFPE tissue is frequently used for somatic mutation testing, the possibility that a rare mutation represents a procedural artifact must be considered. Furthermore, as especially relevant to testing in NSCLC, using extremely limited tissue and DNA quantities, as may be seen with some small specimens, can also lead to sequencing artifacts [98]. Thus, rare mutations must be interpreted with these procedural artifacts in mind, especially for genes which have been extensively characterized. Rare mutation findings should be repeated from the earliest feasible stage of testing and potentially with an alternate testing method. If a rare mutation is confirmed, the report should reflect that the clinical implications are not well understood, unless the scientific medical literature supports an interpretation of clinical significance. Public databases for interpretation of cancer gene mutations include the Catalogue of Somatic Mutations in Cancer (COSMIC) [99] and dbSNP through NCBI. These resources are especially valuable in identifying literature relevant for the interpretation of a specific mutation.
For gene mutations which are previously reported with a known clinical significance, the molecular pathology report should include an interpretive statement as to the clinical relevance of the finding. The implications of a particular result with respect to prognosis or responsiveness to a targeted therapy can change rapidly with new clinical studies, and efforts should be undertaken to ensure the most up-to-date clinically validated implications are noted.
Gene Rearrangement Assays
FISH
Usually, interpretation of FISH results using the ALK BA probe is straightforward. However, the fact that the EML4 and ALK genes are separated by a small distance (12.8 MB) makes the identification of split signals subtle and challenging in a fraction of tumor cells. In the native copy of ALK, the orange and green probes recognize homologous sequences that are physically close to each other and show up as a fused orange (red)/green fluorescent signal (Fig. 32.3a). Therefore, fusion fluorescent signals in this setting indicate a normal copy of the ALK gene. When the break occurs around the ALK intron 19, the orange (red) signals from the 3′ probe sequences separate from the green signals of the 5′ probe sequences (Fig. 32.3b). When EML4-ALK fusion occurs, the sequences recognized by the green probe are positioned approximately 12.8 MB more centromeric to their original locus. Because of the relatively small genomic distance between these signals, scoring criteria require that the split seen be at least two signal diameters wide. In approximately 30 % of the rearrangements involving ALK, chromosomal deletions also occur in association with the inversion causing loss of the genomic sequences recognized by the green-labeled 5′ end sequences (Fig. 32.3c). Thus, the split between orange (red) and green signals and the presence of single orange (red) signals are interpreted as a rearrangement of the ALK gene. A minimum of 50 cells should be counted for assay scoring, and when greater than 15 % of cells are scored as positive (either split pattern or single orange (red) pattern), the result is interpreted as positive for an ALK rearrangement. Additional scoring criteria for the FDA-approved companion diagnostic include an equivocal range of percent positive cells with requirement for a second reader in such cases, although a routine two-reader practice is recommended.
Reverse-Transcription PCR
Interpretation of RT-PCR assays for the detection of ALK rearrangements has particular benefits in that it provides the most direct evidence of a gene rearrangement. Furthermore, this technique can provide the details not only of the 5′ gene fusion partner identity in each case but also the specific exon fusion (Fig. 32.4). In ALK gene-rearranged NSCLC, there are at least three different 5′ gene fusion partners (EML4, KIF5B, and TFG) and multiple different fusion variants in which different exons of EML4 (and occasionally ALK) have been detected for EML4–ALK gene fusions [32–34, 100, 101]. Distinction of which specific fusion partner is involved in the rearrangement can be accomplished through size analysis for some assays, wherein specific fusions produce products of predictable size or may require sequencing of the fusion gene product. For assays which identify fusion events based on the presence of an amplicon of specific size, sequencing should be routinely performed on the amplification product to ensure that the amplicon detected is actually a gene fusion transcript and not the results of false priming. If the assay is designed in a directed fashion (i.e., uses bidirectional primers for the detection of specific fusion variants), negative results must always be interpreted in the context of known fusion partners not detected by the assay and also consider false-negatives attributable to novel fusion partners not yet described. Additionally, as these assays are based on RNA which is typically derived from FFPE tissue, consideration of false negatives attributable to low RNA quality is imperative, and appropriate quality control metrics (such as independent measures of RNA quality) should be included and evaluated for assays of this nature. If such quality metrics are suboptimal, reporting should include caveats about potential false negatives. Finally, this technique has the potential to be extremely sensitive because of the combined effects of PCR amplification and the lack of background signal as only the fusion gene transcript is detected by this assay.
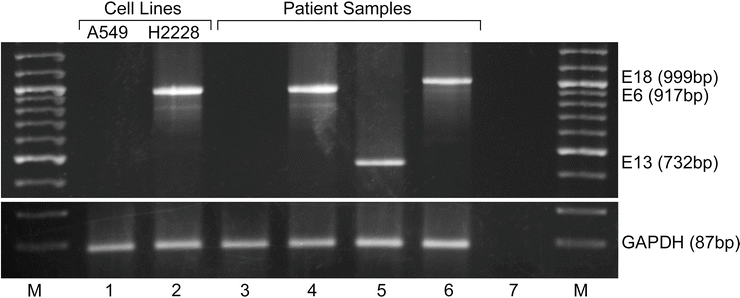
Figure 32.4
Reverse-transcription PCR (RT-PCR) for the detection of known ALK fusions from frozen non-small cell lung cancer (NSCLC) tissue. Multiplex RT-PCR including forward primers to exons 2 and 13 of EML4 and one reverse primer to exon 20 of ALK are utilized in a single reaction. Lanes 1 and 2 are results for negative (A549) and positive (H2228) cell lines, respectively. Lane 3 is a patient sample negative for ALK rearrangement. Lanes 4–6 are patient samples with ALK rearrangements with different EML4 breakpoints. Lane 7 is a no template control. GAPDH mRNA control reactions are shown in the lower panel. Negative results from an assay with amplicons >200–250 bp should be interpreted with caution, and multiplex primers can be designed with amplicon size aimed for detection in FFPE [31, 97]. M molecular weight markers
Gene Amplification Assays
Interpretation of results in FISH assays for copy number analysis is relatively simple. The number of copies per cell of a probe for the target gene sequence is compared with the number of copies of a reference probe, commonly represented by the centromere of the chromosome containing the target gene. Other gene sequences mapped in the same chromosome arm or in the opposite arm of the target gene are acceptable as reference sequences and sometimes even preferable, for instance, when complex rearrangements involve loss of centromeric sequences. Gene amplification is determined when the copy number ratio between the target and reference probe signals is greater than a prespecified value, commonly 2.0.
When a target gene is located close to the chromosomal centromere, both the target gene and the centromeric region may be amplified, making interpretation using a ratio greater than 2.0 inappropriate. EGFR maps at 7p12 near the centromere and is amplified in approximately 10 % of NSCLC [102]. With EGFR gene amplification, the chromosome 7 centromeric sequences also can be amplified, in which case the ratio between the mean EGFR probe signals per cell and the mean centromere 7 probe signals per cell will be approximately 1. Customized scoring systems have been developed to account for such phenomenon, when more than two probe signals for both the EGFR gene and the centromere 7 are identified [103].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


