Figure 28.1
Tumor spectrum in individuals affected with LFS or LFL. The proportion of specific types of tumors among a total of 822 tumors reported in patients with LFS or LFL features is illustrated in the bar graph. Adapted from the International Association for Research on Cancer (IARC) database (R17, November 2013, http://p53.iarc.fr/)
LFS is an autosomal dominant disease. This means inheritance of a single copy of the mutated gene predisposes an individual to the disease. Based on clinical presentation, two forms of LFS are recognized: classic Li-Fraumeni syndrome (LFS) and Li-Fraumeni-like syndrome (LFL). Classic LFS is defined by the following criteria: a proband with a sarcoma diagnosed before 45 years of age, a first-degree relative with any cancer under 45 years of age, and a first- or second-degree relative with any cancer under 45 years of age or a sarcoma at any age [1]. LFL is characterized by a proband with any childhood cancer or sarcoma, brain tumor, or adrenal cortical tumor diagnosed before 45 years of age, a first- or second-degree relative with a typical LFS cancer (sarcoma, breast cancer, brain tumor, adrenal cortical tumor, or leukemia) at any age, and a first- or second-degree relative with any cancer under the age of 60 years [3]. A second definition of LFL is that the affected person can have two first- or second-degree relatives with LFS-related malignancies at any age [4]. The classic LFS criteria were refined and broadened to facilitate identification of individuals without immediate family history.
Breast cancer, brain tumor, and soft-tissue sarcoma account for more than 50 % of the tumors reported in patients with LFS or LFL features (Fig. 28.1) [5, 6]. Meanwhile, individuals with LFS are more likely to develop cancer at younger ages. Age-specific penetrance for cancer in LFS is about 50 % by age 30 years and 90 % by age 60 years. For individuals with LFS carrying germline mutations in the TP53 gene, the median age of cancer onset can be even earlier [7–9]. In contrast, only 2 % of cancers occur under the age of 30 years in the general population [10]. Genetic testing is recommended for patients with multiple tumors, two of which represent characteristic LFS tumors diagnosed at age 36 or younger, or patients with adrenocortical carcinoma diagnosed at any age, regardless of family history [11].
Molecular Basis of Disease
Germline mutations in the tumor suppressor gene, TP53, are associated with LFS and LFL. The human TP53 gene is located on chromosome 17p13.1. This gene spans approximately 20 kb on chromosome 17p13 with 11 exons of which exon 1 is not translated. The gene has multiple promoters and can give rise to multiple isoforms by alternate splicing. The TP53 gene encodes the tumor protein p53 (TP53) that plays an important role in cell cycle control and apoptosis. As a DNA-binding protein, TP53 is composed of several functional domains: N-terminal transcription activation domains (TAD, consists of AD1 and AD2), the central DNA-binding domain (DBD), and the C-terminal tetramerization domain. Other important structural elements include the proline-rich domain, the C-terminal regulatory domain, and several nuclear localization or export signals. The central DNA-binding domain is crucial to the TP53 protein-DNA interaction and is the most frequent site for mutations, especially at codons 248, 273, and 213 (Fig. 28.2). Most of the posttranslational modification sites are localized in the C-terminal or N-terminal domains, which regulate the stability and activity of TP53 [12].
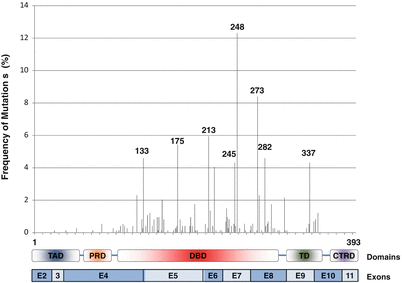
Figure 28.2
Structural organization of the coding exons of the TP53 gene and functional domains of TP53 are illustrated at the bottom of the figure. Relative frequencies and codon positions of germline TP53 mutations found in LFS and LFL families are plotted. Only single base substitutions and insertions/deletions in the codons are listed. Adapted from the International Association for Research on Cancer (IARC) database (R17, November 2013, http://p53.iarc.fr/)
The TP53 gene is regarded as the guardian of the genome and is the most extensively studied tumor suppressor gene. In response to diverse cellular stresses, TP53 regulates a wide variety of genes involved in various cellular processes, including apoptosis, cell cycle, DNA damage repair, senescence, and cellular metabolism. In response to DNA damage caused by ionizing radiation or ultraviolet (UV) light, TP53 protein is stabilized by dissociation from its negative regulator, murine double minute 2 (MDM2). TP53 then translocates to the nucleus and activates the expression of relevant downstream genes, especially CDKN1A (CIP1–WAF1) [13]. CDKN1A protein can function as a cyclin-dependent kinase inhibitor and mediate the TP53-dependent G1-S cell cycle arrest through targeting cyclin-dependent kinases, such as CDK2 and CDK4. The activation of the cell cycle checkpoint facilitates the repair of damaged DNA [14].
Somatic TP53 mutations occur in almost every type of cancer at varying frequencies, and they are more frequent in advanced stage or in more aggressive tumor types [15]. The International Agency for Research on Cancer (IARC) maintains a regularly updated database of all known TP53 gene variations and mutations (germline and somatic) identified in human tumor samples (http://TP53.iarc.fr/). As of November 2013, the R17 release of IARC TP53 database contains 750 germline mutations, 28,000 somatic mutations, and TP53 mutation status of 2,700 cell lines. The database also includes functional data of 2,314 mutant TP53 proteins. The codon distribution and relative frequencies of germline mutations in TP53 as observed in LFS or LFL families are summarized in Fig. 28.2. The spectrum of different types of TP53 mutations observed in LFS and LFL is summarized in Fig. 28.3 (R17, November 2013) [5, 6, 16].
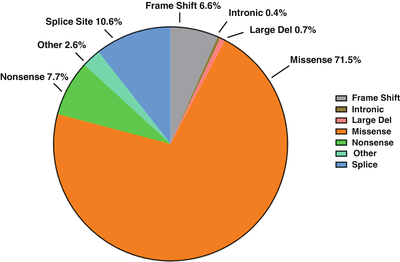
Figure 28.3
Types of germline mutations in the TP53 gene found in LFS and LFL cases. Pie chart shows the percentage of the different types of observed mutations. Adapted from the International Association for Research on Cancer (IARC) database (R17, November 2013, http://p53.iarc.fr/)
The majority of the germline mutations (83.7 %) reside within the central DBD, which can interfere with the ability of TP53 to bind to its target DNA sequences. Common mutations found in LFS and LFL families occur in codons 175, 213, 245, 248, 273, and 337 and account for 39 % of all the single base substitutions. Many tumor suppressor genes like RB1 and APC are mainly altered by nonsense or truncating mutations. In contrast, the majority of germline TP53 mutations found in LFS and LFL families are missense substitutions (71.5 %). Other alterations include splice site mutations (10.6 %), nonsense mutations (7.7 %), frameshift mutations (6.6 %), and other infrequent alterations such as large deletion (<1 %) and deep intronic mutations (<1 %) (Fig. 28.3) [5, 6]. Heterozygous deletion of the entire TP53 gene has been observed in one of 79 LFS families without any coding sequence mutation in TP53 [17]. Attwooll and colleagues detected a single nucleotide deletion within the TP53 promoter region in two of 18 LFS/LFL families without an identifiable TP53 coding sequence mutation. Later, their data indicated that this mutation does not have any functional consequence and is a rare polymorphism [18].
The results of sequence analysis of the entire coding region of TP53 indicate that 60–80 % classic LFS families carry germline TP53 mutations, while only 8–22 % of individuals with LFL have detectable mutations [19]. The frequency of de novo mutations in LFS is approximately 7–20 % [8, 9]. The reasons for a lower detection rate of TP53 mutations in LFS families include undetected intronic or cytogenetic lesions of the TP53 gene, other genes predisposing to LFS, or the presence of mutations in modifier genes.
The variability of the cancer spectrum and age of onset within LFS families suggests the presence of modifier genes. One of the plausible candidate genes is MDM2, which is a negative regulator of TP53 through its E3 ubiquitin ligase activity [20]. A single nucleotide polymorphism (SNP) within the promoter region of MDM2, SNP309 T>G (dbSNP ID, rs2279744), has been associated with earlier onset of tumors among germline TP53 mutation carriers [21, 22]. It has also been suggested that there is a cumulative effect in cases where both MDM2-SNP309 polymorphism and TP53 codon 72Arg variant (R72P, dbSNP ID, rs1042522) are present in LFS patients [21, 22]. A different line of evidence from Tabori and colleagues suggested the presence of accelerated telomere attrition in LFS families and postulated that the shorter telomere length in TP53 mutant carriers can predict genetic anticipation observed in LFS patients and serves as a rational biological marker [23].
The relatively low frequency of detectable germline mutations in TP53 in many LFS and LFL probands suggests the possible involvement of other genes, which may or may not directly involve in the TP53 pathway. The candidates include but are not limited to p63, p73, BCL10, BAX, CDKN2A, PTEN, BRCA1, BRCA2, CHEK1, and CHEK2 genes. However, despite numerous efforts to screen these genes for mutations, studies have not detected any high-penetrance mutations [19, 24, 25]. For example, 1100delC and I157T mutations in the CHEK2 gene have been recurrently found in LFS families and possibly represent low-penetrance mutations, which confer increased risk for breast, prostate, and thyroid cancer [19].
It has been generally accepted that mutation in all tumor suppressor genes will act in a recessive manner, which means both alleles should be inactivated to cause tumor initiation and progression [26]. Although the majority of TP53 mutations are missense mutations that reside within the central DNA-binding domain, loss of heterozygosity is only identified in approximately 60 % of tumors [27, 28]. Approximately two-thirds of the tumors arising in LFS patients carry missense mutations on one allele of TP53 while the other TP53 allele remains as wild type. One explanation is that the mutant form of TP53 can exert a dominant negative effect and functionally inactivate the wild-type TP53. Various lines of evidence further indicate that certain TP53 mutations may result in new TP53 functions that contribute to tumor initiation and progression and are referred to as gain-of-function mutations [29].
Clinical Utility of Testing
According to the practice guidelines in oncology (v.1.2010, of National Comprehensive Cancer Network (NCCN) http://www.nccn.org/professionals/physician_gls/f_guidelines.asp), TP53 gene testing is considered necessary for individuals with a known family history of a TP53 mutation, or with a suspected or known clinical diagnosis of LFS or LFL, or with a diagnosis of early-onset breast cancer before 30 years with a negative BRCA1 and BRCA2 test. Testing also is recommended if there is a family history of sarcoma, brain tumor, or adrenocortical carcinoma. Several general recommendations and guidelines are available regarding diagnostic testing for germline TP53 mutations among cancer-prone individuals [30]. Individuals chosen for genetic testing on the basis of their family histories should be given current, relevant information on the test to make an informed voluntary decision.
Since cancers occur with high frequency among children in LFS families, testing these children (rather than delaying testing until young adulthood) is recommended. However, as children mature, obtaining of their assent or dissent to testing is appropriate, as well as the consent of their parents. Annual surveillance strategies are recommended for at-risk children and include complete physical examination, urinalysis, blood count, and abdominal ultrasound examination. Additional organ-targeted surveillance based on family history (e.g., imaging studies of the head if a relative had a childhood brain tumor) also is recommended.
For women with early-onset breast cancer with a negative BRCA1 and BRCA2 gene mutation, TP53 gene testing is only meaningful in the context of a family history characteristic of classic LFS or LFL criteria. Several studies have shown that the likelihood of a germline TP53 mutation in this population can range from 0–7 %, even if no family history of cancer is present [8, 9, 31, 32]. Females with a germline TP53 mutation have the option of prophylactic mastectomy to reduce the risk of breast cancer [33]. Routine mammograms and clinical breast examinations are effective in women over age 40 years, but have not been proven to be beneficial for younger women with LFS or LFL. However, controversy exists regarding the use of routine mammograms in women with LFS, because of possible radiation sensitivity associated with TP53 mutations [10, 34]. In addition, based on family history of specific cancers, colonoscopies or full-body MRI examination or PET scan has been suggested
Available Assays
Several approaches to clinical testing for TP53 are used, but most use sequence analysis due to the distribution of mutations throughout the gene. A comprehensive list of available clinical tests is available through the Genetic Testing Registry database (http://www.ncbi.nlm.nih.gov/gtr/).
For sequence analysis, PCR amplification of the entire coding region of TP53 (exons 2–11) and the immediate flanking intronic sequences is performed on genomic DNA isolated from blood or saliva of the proband(s). Direct sequencing is performed using traditional automated fluorescent dideoxynucleotide sequencing. Approximately 75 % of disease-causing mutations can be identified in exons 5–8 of the TP53 gene by sequence analysis [27]. Identified sequence variations can be classified as pathogenic mutations, benign variants, or variations of unknown clinical significance. Splice site mutations are common (12 %) in LFS and LFL probands [5, 6]. Further interpretation of the results of molecular analysis is usually based on the clinical presentation and family history of the patient.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


