Figure 6.1
Common types of chromosome abnormalities detected by standard chromosome analysis (G-banded karyotyping). (a) Aneuploidy, defined as the loss or gain of an entire chromosome, shown as trisomy 21. (b) Terminal deletion, shown as a deletion of distal 11q (normal chromosome 11 on left, with deleted region bracketed, and deleted 11 on right). (c) Interstitial deletion, shown within 3p (normal chromosome 3 on left, with deleted region bracketed, and deleted 3 on right). (d) Inversion, shown as a pericentric inversion of chromosome 7 (normal chromosome 7 on left with inverted region bracketed, and inverted 7 on right). (e) Isochromosome, defined as the loss of one chromosome arm with duplication and mirror image of other arm, shown as isochromosome Xq (normal X on left and isochromosome Xq on right). (f) Balanced translocation, shown between 10q and 12q (with normal 10 and 12 on left of each pair and translocated 10 and 12 on right with arrows marking breakpoints of the translocation). (g) Unbalanced translocation, shown between 14q and Xq with two normal 14 chromosomes, one normal X, and one X with a deletion of part of Xq and replacement with gain of 14q region bracketed, leading to partial 14q trisomy and partial Xq monosomy.
Fluorescence In Situ Hybridization
FISH approaches were first described in 1986 [12] and have become essential for many comprehensive cytogenetic analyses. Most any DNA segment can be used as a FISH probe. For clinical use, most probes are between 100 and 500 kb. This provides a resolution far greater than that of G-banding for the identification of deletions, insertions and translocation breakpoints. The types of probes include (1) painting probes, which are used to identify the chromosomal origin of a DNA segment and are useful for distinguishing translocations; (2) centromere probes that hybridize to alpha-satellite DNA located at the centromere of each chromosome, which are used to identify aneuploidy of specific chromosomes or to identify the chromosomal origin of marker chromosomes; (3) beta-satellite probes, which are used to identify beta-satellite regions on the short arm of acrocentric chromosomes; (4) subtelomeric probes, which are used to detect subtelomeric rearrangements of chromosome arms; and (5) locus-specific probes, which hybridize to unique gene sequences and are used to identify contiguous gene syndromes and other microdeletion or microduplication syndromes (Fig. 6.2).
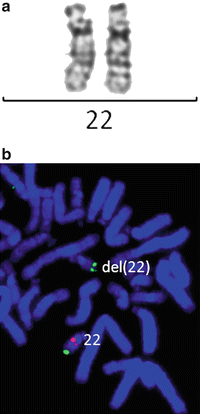
Figure 6.2
Chromosomally cryptic 22q11.2 deletion detected by FISH, but not by G-banded karyotyping. (a) Normal banding pattern for each chromosome 22. (b) FISH with a specific probe to the TUPLE1 gene in red and control probe in green show a deletion of one copy of this gene on one chromosome 22
FISH allows the determination of the frequency and location of specific DNA sequences in cells in any state of the cell cycle and in archived formalin-fixed, paraffin-embedded tissues. However, FISH can only detect deletions, duplications, and rearrangements of the chromosomal region complementary to the probe. Clinically, suspicion of a syndrome with known cytogenetic etiology is normally required to run a FISH study. Also, the number of probes that can be mixed and used in a single FISH assay is limited and depends on the number of fluorochromes available for probe labeling; in most clinical applications, this is limited to three unique regions probed per hybridization.
Chromosomal Microarray Analysis
Chromosomal Microarray Methodology
CMA allows for the detection of loss or gain at multiple loci at a much higher resolution than either conventional chromosome analysis or FISH. For CMA testing using aCGH, the patient’s genomic DNA and control genomic DNA are labeled with different fluorochromes and co-hybridized onto DNA substrates immobilized on a solid support (Fig. 6.3). Several aCGH platforms have been developed for diagnostic purposes. Initially, bacterial artificial chromosomes (BACs, between 80 and 200 kb) were used as array targets (Fig. 6.4) [13]. Oligonucleotide arrays (25–85 bp) were subsequently constructed to allow for higher resolution [14, 15].
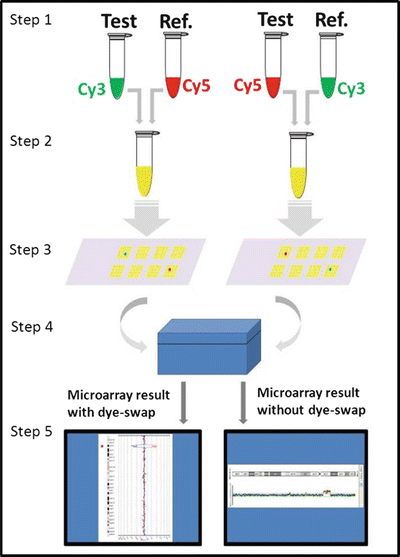
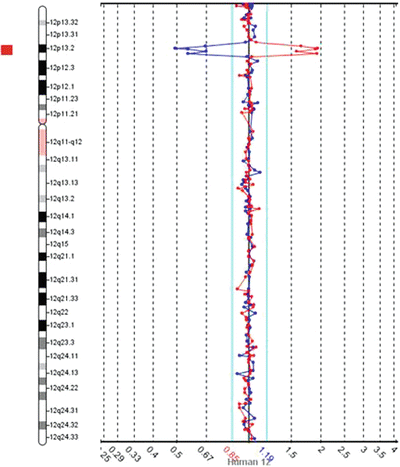
Figure 6.3
Steps in aCGH procedure. Step 1: Test and reference DNA are fragmented and labeled with fluorescent dyes, Cy3 and Cy5. Step 2: Equal amounts of labeled test and reference DNAs are mixed together. Step 3: The combined labeled test and reference DNAs are applied to the microarray and compete to hybridize to the target DNA (segments of the human genome) on the microarray. Step 4: Fluorescent signals are measured in the microarray scanner. Step 5: Analysis software generates data linking signal to relative copy number of test DNA compared to reference DNA for each probe on the microarray. To confirm the result, this process is repeated with the fluorochromes switched between the test and reference DNAs (called a dye-swap), so that a true loss or gain would have the opposite fluorescent signal in the repeat test. In lieu of a dye-swap repeat, multiple independent probes adjacent in the genome but separate on the microarray showing a contiguous loss or gain increases confidence in the accuracy of result.
Figure 6.4
Example of a deletion detected by the original aCGH with the dye-swap repeat using BAC-based targets. Data from chromosome 12 with each dot representing a BAC probe mapping approximately every one megabase across chromosome 12. In both runs, less of the labeled test DNA hybridized to the BACs in region 12p13 when compared to the reference DNA, as represented by the reciprocal deviation from the central black line (representing zero) on the Y-axis and the red bar to the left of the schematic chromosome 12 (for this software, when equal hybridization of test and reference DNAs occurs, the dots map within the aqua lines near the central black line)
Arrays that target clinically meaningful regions of the genome have been used for postnatal and prenatal cases with normal chromosome analysis results and for general screening purposes [16–20]. The whole genome designed array offers full coverage of the genome as the technology and genomic architecture allows. The copy number of some regions of the genome cannot be accurately evaluated by this technology, notably repetitive regions and small deletions and duplications. Whole-genome design is important for the discovery of new chromosomal syndromes [21, 22] and detection of recently discovered syndromes that may have occurred since the design of a targeted array. Many platforms in clinical use are currently a combination of both whole- genome coverage and enrichment of targets at known clinically relevant regions.
The SNP array is another type of oligonucleotide array. SNPs are DNA sequence variations in which a single nucleotide in the sequence of the oligonucleotide differs between individuals or between paired chromosomes in an individual. Similar to aCGH, SNP arrays contain probes that can detect imbalances at thousands of loci in the genome. In contrast to aCGH, SNP arrays also can be used for detecting long contiguous stretches of homozygosity, which may be caused by either consanguinity or uniparental disomy (UPD) of a genomic interval (Fig. 6.5). Both consanguinity and UPD increase the risk of autosomal recessive conditions. In addition, UPD of specific chromosomal regions is associated with imprinting defects, such as UPD of chromosome 15 in patients with Prader-Willi syndrome and Angelman syndrome.
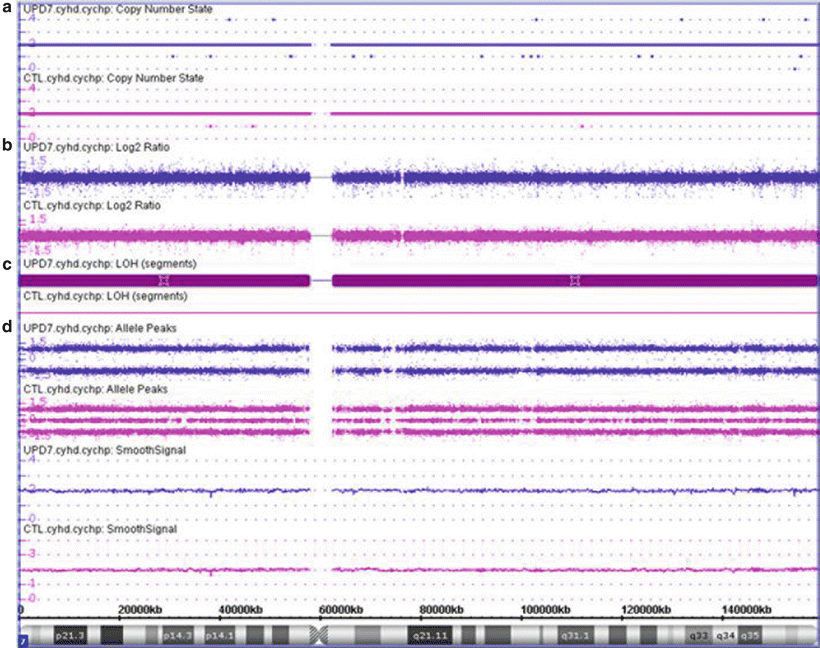
Figure 6.5
CMA finding consistent with uniparental isodisomy of chromosome 7. UPD7 case shown in purple and a normal case shown in pink for comparison. (a) The copy number state is determined to be 2 (normal diploid) across the vast majority of the chromosome both for the UPD7 and normal cases (CTL). (b) Estimation of the copy number state based on the log2 ratio of the hybridization of the patient DNA to the control DNA, with signal centered at zero. (c) Loss of heterozygosity is noted across the entire chromosome for the UPD7 case (maroon bar) due to the allele peak pattern which shows only values at 1.0 and −1.0; whereas the normal case shows normal heterozygosity (no maroon bar) with values at 1.0, 0, and −1.0. These values are determined by assigning each allele a value of 0.5 and then plotting A–B. Therefore a locus with AA is (0.5 + 0.5) − 0 = 1; a locus with AB is 0.5 − 0.5 = 0; and a locus with BB is 0 − (0.5 + 0.5) = −1. (d) For the UPD7 case, all of chromosome 7 shows no evidence of any AB loci, suggestive that both chromosome 7 s are 100 % identical. Maternal UPD7 is associated with Russell-Silver syndrome; whereas paternal UPD7 has not been associated with a specific clinical consequence. Additionally, recessive disorders mapping to chromosome 7, such as cystic fibrosis, should be considered for this child. Although this example shows isodisomy for all of chromosome 7, also note that UPD may have parts of the affected chromosome showing isodisomy whereas other parts of the same chromosome still show heterodisomy due to crossing over between the homologous chromosomes.
Advantages of CMA Testing
Compared with G-banded karyotyping, CMA analyzes chromosomes for genomic gains and losses at a much higher resolution, which allows the detection of smaller or cryptic abnormalities (Figs. 6.6 and 6.7) and provides better definition of cytogenetic abnormalities (Fig. 6.8). The detection of abnormalities by CMA is usually more detailed, automatable, and less subjective than G-banded karyotyping. Furthermore, archived or non-growing tissue can be used for CMA, but not G-banded karyotyping. CMA also may provide information on the mechanism of the genetic aberration. For example, depending on the observed pattern of imbalance in the proband and the parents, the proband may be predicted to have an unbalanced segregation of a translocation or inversion which is present in a balanced form in either parent. Also, analysis of the flanking regions of the imbalance allows determination of whether the rearrangement is mediated by non-allelic homologous recombination [25]. Information about the likely mechanism then informs the likelihood of recurrence in subsequent pregnancies.
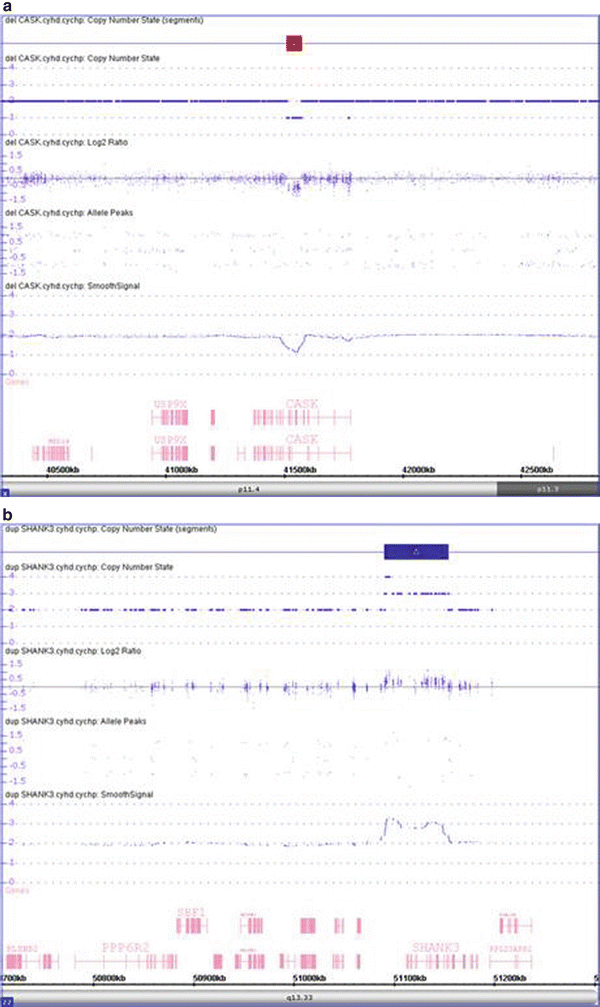
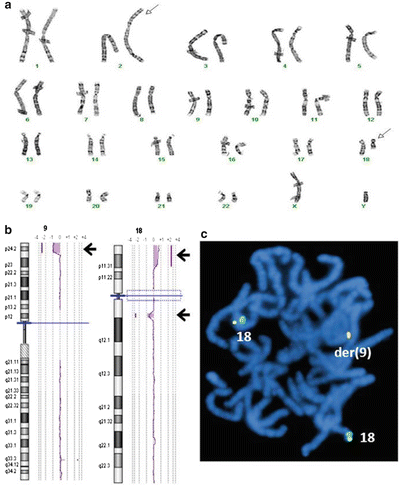
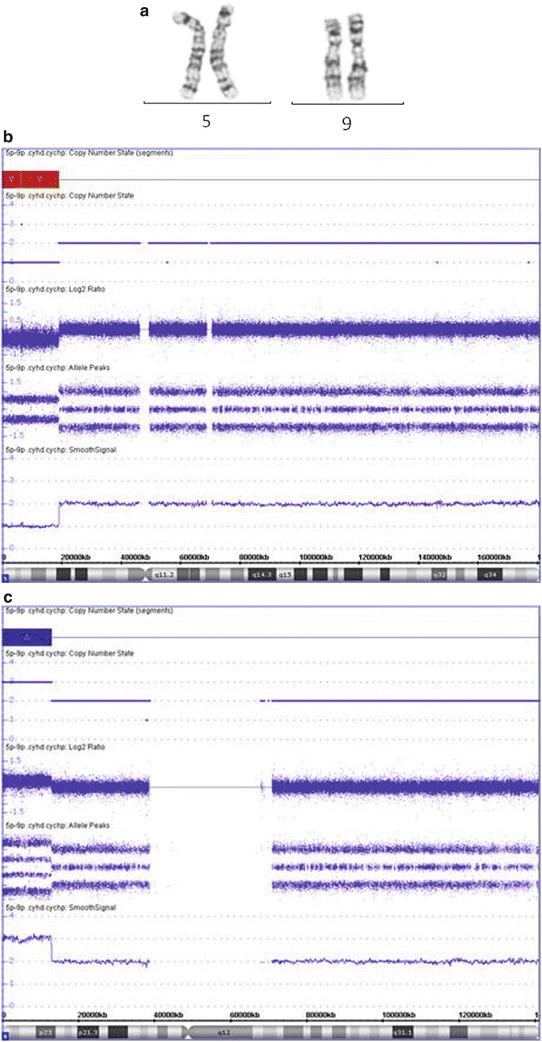
Figure 6.6
Detection of intragenic loss and small gain using CMA. (a) The CMA shows a 63 kb deletion within Xp11.4 in a female patient (region highlighted by red bar at top). This deletion involves three exons of the gene CASK. Deletions of CASK have been found in female and male patients with X-linked cognitive disability, optic atrophy, brainstem and cerebellar hypoplasia, microcephaly, and dysmorphic facial features [23]. (b) The CMA shows a 64 kb gain within 22q13.33, indicating trisomy for this region (region highlighted by blue bar at top). This duplication has a breakpoint within the SHANK3 gene. Disruption of SHANK3 has been reported in patients with global developmental delay [24].
Figure 6.7
Identification of cryptic abnormality by CMA in addition to a visible chromosome abnormality by G-banded karyotype. (a) Chromosome G-banding shows a translocation between 2q and 18q (arrows). (b) Genomic microarray shows a 1.2 Mb interstitial deletion at the 18q breakpoint, but also a 5.9 Mb terminal deletion of 9p and a 6.0 Mb terminal duplication of 18p (arrows). (c) FISH with the 18p subtelomere probe (fusion signal) shows additional signal is on the derivative 9, consistent with an unbalanced translocation not detected in chromosome analysis
Figure 6.8
CMA often provides a better definition of a chromosome abnormality, leading to better genotype-phenotype correlation and recurrence risk estimation. (a) Chromosome analysis shows a terminal deletion on the short arm of chromosome 5 (abnormal chromosome on right) with two normal appearing chromosome 9 s. (b and c) The CMA analysis indicates that there is a 19.0 Mb terminal deletion of 5p (b), and a 12.8 Mb terminal duplication of 9p (c), suggesting an unbalanced translocation involving 5p and 9p. This abnormality is associated with the Cri-du-Chat syndrome and 9p duplication syndrome separately, so this patient is likely to have a clinical phenotype with features of both syndromes. In addition, there is a higher probability of a parent carrying a balanced rearrangement of 5p and 9p then if this was only a 5p deletion. This increases the urgency for parental testing for recurrence risk determination.
The diagnostic yield of a G-banded karyotype in individuals with ID/DD is approximately 3 %. FISH analysis of subtelomeric regions may provide a diagnosis in another 2–3 % of cases [26]. Several large cohort studies have shown that CMA has the highest yield as a single diagnostic test for individuals with ID/DD [5, 11, 27–29]. The detection rate varies between studies, likely due to differences in platform design, populations studied, and interpretation methods (see below); however, these studies still demonstrated between a 10 and 28.9 % detection rate for pathogenic aberrations in patients with ID/DD, and less than 0.6 % of cases with probable disease-causing balanced de novo aberrations remained undetectable. Thus, CMA has been recommended as a “first-line test” in the initial evaluation of patients with nonsyndromic ID/DD by the American College of Medical Genetics (ACMG) Practice Guidelines [10].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


