Dalia S. Nagel and Helen M. Shields

A peptic ulcer is a break in the mucosa of the stomach (gastric ulcer) or duodenum (duodenal ulcer). Four-and-a-half million people in the United States suffer from active peptic ulcer disease, and 500,000 new cases of peptic ulcer disease are diagnosed each year. The lifetime prevalence of peptic ulcer disease is approximately 10%, and the estimated annual cost for treatment exceeds $1 billion.
There are several pathophysiologic mechanisms for peptic ulcer disease, and clinical management often requires multiple pharmacologic strategies. This chapter describes the physiology of gastric acid secretion and the pathophysiology underlying the formation of peptic ulcers. The pharmacologic agents used in the treatment of peptic ulcer disease are then discussed in relation to the pathophysiology that is interrupted by these drugs.
 Tom is a 24-year-old graduate student. He is in good health, although he smokes approximately two packs of cigarettes and drinks five cups of coffee a day. He is currently under stress because of the impending deadline for his computer science thesis.
Tom is a 24-year-old graduate student. He is in good health, although he smokes approximately two packs of cigarettes and drinks five cups of coffee a day. He is currently under stress because of the impending deadline for his computer science thesis.
For the past 2 weeks, Tom has noted a burning pain in his upper abdomen that occurs 1–2 hours after eating. In addition, the pain frequently awakens him at approximately 3:00 AM. His pain is usually relieved by eating and by taking over-the-counter antacids.
When the pain increases in intensity, Tom decides to visit his internist, Dr. Smith, at University Health Services. Dr. Smith notes that the abdominal examination is normal except for epigastric tenderness. Dr. Smith discusses diagnostic options with Tom, including an upper gastrointestinal x-ray series and an endoscopic examination. Tom chooses to undergo the endoscopic examination. During the examination, an ulcer is identified in the proximal portion of the duodenum on the posterior wall. The ulcer is 0.5 cm in diameter. A mucosal biopsy of the gastric antrum is performed for detection of Helicobacter pylori.
Tom is diagnosed with a duodenal ulcer. Dr. Smith prescribes omeprazole, a proton pump inhibitor. The next day, when the pathology report indicates the presence of an H. pylori infection, Dr. Smith prescribes bismuth, clarithromycin, and amoxicillin in addition to the proton pump inhibitor. Dr. Smith also advises Tom to stop smoking and drinking coffee.
Questions
1. What risk factors did Tom have for the development of peptic ulcer disease? What is the role of H. pylori in this disease?
2. Why was Tom given clarithromycin rather than metronidazole for treatment of his H. pylori infection?
3. Why was Tom also treated with a proton pump inhibitor?
 Marianne is a 54-year-old administrator in a printing shop who types 4–5 hours a day. She develops carpal tunnel syndrome and begins to take several aspirin daily for the pain. One month later, Marianne develops a burning pain in her upper abdomen. After vomiting “coffee grounds” material and noticing that her bowel movements are black, she decides to visit the emergency room of her local hospital. The on-call gastroenterologist performs an endoscopy and confirms that Marianne has a gastric ulcer that has recently bled. The gastroenterologist explains to Marianne that she has a peptic ulcer. Marianne’s breath test is negative for H. pylori, and she is told that aspirin is the most likely cause. Marianne is treated with antacids and ranitidine (an H2 receptor antagonist) and is told to stop taking nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin. The gastroenterologist reviews with Marianne the list of pain-relieving medications that are considered to be NSAIDs.
Marianne is a 54-year-old administrator in a printing shop who types 4–5 hours a day. She develops carpal tunnel syndrome and begins to take several aspirin daily for the pain. One month later, Marianne develops a burning pain in her upper abdomen. After vomiting “coffee grounds” material and noticing that her bowel movements are black, she decides to visit the emergency room of her local hospital. The on-call gastroenterologist performs an endoscopy and confirms that Marianne has a gastric ulcer that has recently bled. The gastroenterologist explains to Marianne that she has a peptic ulcer. Marianne’s breath test is negative for H. pylori, and she is told that aspirin is the most likely cause. Marianne is treated with antacids and ranitidine (an H2 receptor antagonist) and is told to stop taking nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin. The gastroenterologist reviews with Marianne the list of pain-relieving medications that are considered to be NSAIDs.
Two weeks pass. Marianne informs her gastroenterologist that the pain in her wrist has become unbearable and that she must continue taking aspirin to be able to type at work and keep her job. The gastroenterologist tells Marianne that she can take aspirin as long as she switches her antiulcer medication from an H2 antagonist to a proton pump inhibitor.
Questions
4. Why was an H2 antagonist prescribed for Marianne’s ulcer, and why was her medication switched to a proton pump inhibitor when she insisted on using aspirin?
 PHYSIOLOGY OF GASTRIC ACID SECRETION
PHYSIOLOGY OF GASTRIC ACID SECRETION
Neurohormonal Control of Gastric Acid Secretion
Hydrochloric acid is secreted into the stomach by parietal cells, which are located in oxyntic glands in the fundus and body of the stomach. The parietal cell actively transports H+ across its apical canalicular membranes via H+/K+ ATPases (proton pumps) that exchange intracellular H+ for extracellular K+. Three neurohormonal secretagogues regulate this process: histamine, gastrin, and acetylcholine (ACh). Each of these secretagogues binds to and activates specific receptors on the basolateral membrane of the parietal cell, thereby initiating the biochemical changes necessary for active transport of H+ out of the cell.
Histamine, released by enterochromaffin-like (ECL) cells located in and adjacent to the oxyntic glands and by mast cells in the lamina propria, binds to histamine H2 receptors on the parietal cell. H2 receptor activation stimulates adenylyl cyclase and increases intracellular cyclic adenosine monophosphate (cAMP). In turn, cAMP activates cAMP-dependent protein kinase (protein kinase A [PKA]). PKA phosphorylates and activates proteins responsible for trafficking of cytoplasmic tubulovesicles containing H+/K+ ATPase to the apical membrane of the cell. The H+/K+ ATPase does not pump H+ into the tubulovesicles because the permeability of the vesicular membrane to K+ is low. After fusion of the tubulovesicles with the apical membrane, the availability of extracellular K+ allows the H+/K+ ATPase to pump H+ from the parietal cell into the gastric lumen. Concurrent with the trafficking of cytoplasmic tubulovesicles to the apical membrane, cellular activation mobilizes an apical membrane K+ channel to provide the extracellular K+ for this process (Fig. 47-1).
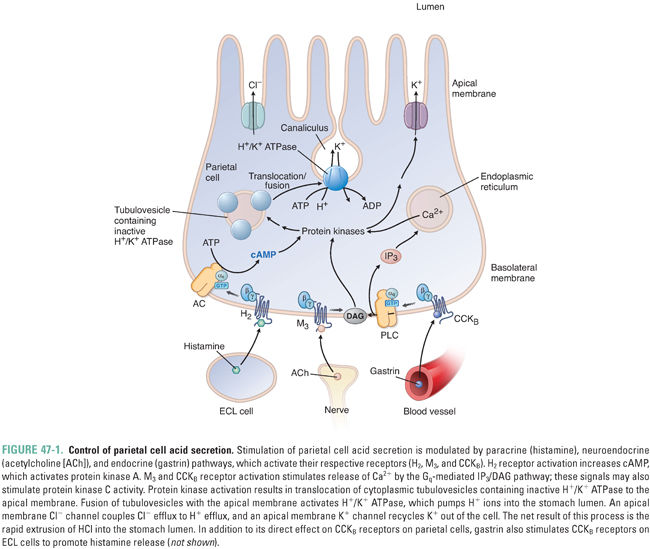
Gastrin is secreted into the bloodstream by G cells in the gastric antrum, and acetylcholine is released from postganglionic nerves with cell bodies located in the submucosa (Meissner’s plexus). Both of these secretagogues bind to their respective G protein-coupled receptors on the parietal cell and thereby activate phospholipase C and increase intracellular calcium levels (Ca2+) (Fig. 47-1). Beyond the involvement of phospholipase C and intracellular Ca2+, the signaling pathways by which parietal cell stimulation by gastrin and ACh leads to H+/K+ ATPase activation remain to be fully elucidated; protein kinase C is likely to be involved. In addition to its relatively minor role in stimulating the parietal cell directly, gastrin has a major role in stimulating the release of histamine by ECL cells (see below).
While histamine, gastrin, and ACh increase acid secretion by parietal cells, somatostatin-secreting D cells and prostaglandins limit the extent of gastric acid secretion. Somatostatin decreases acid secretion via three mechanisms: (1) inhibition of gastrin release from G cells by a paracrine mechanism, (2) inhibition of histamine release from ECL cells and mast cells, and (3) direct inhibition of parietal cell acid secretion. Prostaglandin E2 (PGE2) enhances mucosal resistance to tissue injury by (1) reducing basal and stimulated gastric acid secretion and (2) enhancing epithelial cell bicarbonate secretion, mucus production, cell turnover, and local blood flow.
Phases of Gastric Acid Secretion
Gastric secretions increase considerably during a meal. There are three phases of gastric acid secretion.
The cephalic phase includes responses to sight, taste, smell, and thought of food. “Sham feedings,” experiments in which food is chewed but not swallowed, trigger an increase in acid secretion mediated by vagal stimulation and increased gastrin secretion.
Mechanical distension of the stomach and ingestion of amino acids and peptides stimulate the gastric phase. Distension activates stretch receptors in the wall of the stomach that are linked to short intramural nerves and vagal fibers. Luminal nutrients, such as amino acids, are strong stimulants for gastrin release. Gastrin travels via the blood to the oxyntic mucosa and stimulates ECL cells to release histamine. An important negative feedback on acid secretion in this phase is acid (pH <3)-mediated inhibition of gastrin release from antral G cells. Acid secretion is also inhibited by release of somatostatin from antral D cells.
The intestinal phase involves stimulation of gastric acid secretion by digested protein in the intestine. Gastrin plays a major role in mediating this phase as well.
Factors that protect the gastric mucosa include gastric mucus, prostaglandins (discussed above and in Chapter 43, Pharmacology of Eicosanoids), gastric and duodenal bicarbonate, restitution (repair), and blood flow. The epithelial cells of the stomach secrete mucus, which acts as a lubricant that protects the mucosal cells from abrasions. Composed of hydrophilic glycoproteins that are viscous and have gel-forming properties, the mucus layer enables formation of an uninterrupted layer of water at the luminal surface of the epithelium. Together, the mucus and water layers attenuate potential damage due to the acidic environment of the gastric lumen. Prostaglandins stimulate mucus secretion, whereas NSAIDs and anticholinergic medications inhibit mucus production. In addition, H. pylori disrupts the mucus layer (see below).
Bicarbonate protects the gastric epithelium by neutralizing gastric acid. Bicarbonate is secreted by epithelial cells at the luminal surface of the gastric mucosa, in gastric pits, and at the luminal surface of the duodenal mucosa. Bicarbonate secretion in the duodenum serves to neutralize acid entering the intestine from the stomach.
Restitution refers to the ability of the gastric mucosa to undergo repair. Damage is repaired through migration of undamaged epithelial cells along the basement membrane to fill defects created by the sloughing of injured cells.
The final protective factor is blood flow. Blood flow to the gastric mucosa removes acid that has diffused across a damaged mucus layer.
 PATHOPHYSIOLOGY OF PEPTIC ULCER DISEASE
PATHOPHYSIOLOGY OF PEPTIC ULCER DISEASE
A peptic ulcer is a break in the lining of the stomach or duodenum. The break can involve the mucosa, muscularis mucosa, submucosa, and in some cases, the deeper layers of the muscle wall. This compromise of mucosal integrity can cause pain, bleeding, obstruction, perforation, and even death. Peptic ulcers are caused by an imbalance between protective factors and damaging factors in the gastrointestinal mucosa. This section describes the main pathophysiologic mechanisms involved in ulcer formation, the two most common of which are H. pylori infection and NSAID use.
H. pylori, a Gram-negative, spiral-shaped bacterium, is the most common cause of non-NSAID-associated peptic ulcer disease. H. pylori has been found in the gastric antrum of a significant number of patients with duodenal ulcers and gastric ulcers, including Tom in the introductory case. Eradication of H. pylori leads to lower recurrence and relapse rates in patients with ulcers. The latter finding, together with the fact that many ulcer patients are infected with H. pylori, constitute the major evidence for H. pylori’s causal role in peptic ulcer disease.
H. pylori lives in the acidic environment of the stomach. The initial infection is transmitted by the oral route. Upon ingestion, the microaerophilic bacterium uses its four to six flagellae to move in corkscrew fashion through the gastric mucus layer. H. pylori attaches to adhesion molecules on the surface of gastric epithelial cells. In the duodenum, H. pylori attaches only to areas containing gastric epithelial cells that have arisen as a result of excess acid damage to the duodenal mucosa (gastric metaplasia). H. pylori is able to live in such a hostile environment partly because of its production of the enzyme urease, which converts urea to ammonia. The ammonia buffers the H+ and forms ammonium hydroxide, creating an alkaline cloud around the bacterium and protecting it from the acidic environment of the stomach.
H. pylori’s virulence factors cause damage to the host. Urease is one of these damaging factors because it is an antigen that causes a strong immune response. In addition, ammonium hydroxide produced by urease causes gastric epithelial cell injury. Other virulence factors include lipopolysaccharides (endotoxins), which are components of the bacteria’s outer membrane, as well as a lipase and a protease that are secreted by the bacteria and degrade the gastric mucosa. Cytotoxicity caused by H. pylori has also been linked to two major proteins: cytotoxin-associated gene A (Cag A) and vacuolating cytotoxin (VacA). The cag pathogenicity island is linked to expression of Cag A. This pathogenicity island, which is present in the majority of H. pylori isolates, contains approximately 32 genes that encode a bacterial type IV secretion system. The secretion system inserts into gastric epithelial cells of the host and transports Cag A (and other virulence factors) into the epithelial cells. Once inside the host cell, Cag A undergoes tyrosine phosphorylation by host kinases. Both unphosphorylated and phosphorylated Cag A influence host signaling pathways and host cellular functions, including acid secretion, cytokine release, cellular proliferation and apoptosis, cell polarity, and cell motility. Compared to strains of H. pylori that do not express Cag A, H. pylori strains expressing Cag A have been linked to a higher incidence of duodenal ulcers, gastric ulcers, and gastric cancer.
The persistence of H. pylori can be traced, in part, to the inappropriate immune response that it elicits. Instead of the normal TH2 mucosal immunity response, which controls luminal infections by means of secretory (IgA) antibody, the H. pylori organism elicits a TH1 response. Cytokines associated with the TH1 response induce inflammation and epithelial cell damage.
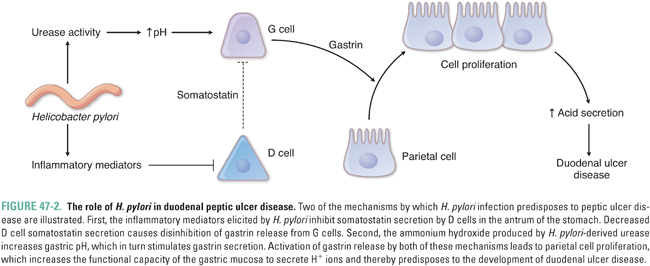
Several additional mechanisms characterize H. pylori-induced peptic ulcer disease (Fig. 47-2). Acid secretion is increased in patients with H. pylori-associated duodenal ulcers. This is thought to result from increased levels of circulating gastrin, causing parietal cell proliferation and increased acid production. Gastrin secretion is elevated by two mechanisms: (1) the ammonia generated by H. pylori produces an alkaline environment near the G cells and thereby stimulates gastrin release and (2) the number of antral D cells is lower than normal in H. pylori-infected patients, resulting in decreased somatostatin production and increased gastrin release. H. pylori also decreases duodenal bicarbonate secretion and thereby weakens the protective mechanisms of the duodenal mucosa.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


