INTRODUCTION
Gout is a uniquely human disease. Most mammals possess uricase, an enzyme that metabolizes purine breakdown products into a freely water-soluble substance, allantoin. Humans, in contrast, excrete most purines as sparingly soluble uric acid. High plasma levels of uric acid can lead to deposition of uric acid crystals in joints, most frequently the first metatarsophalangeal joint (great toe). Acute attacks of gout cause intense pain but typically occur infrequently.
A number of rational therapies exist for the treatment of gout. These therapies are broadly divided into two groups: those that treat acute gout attacks and those that prevent recurrent attacks. Drugs that suppress the immune response to crystal deposition or limit the extent of inflammation may be used for both indications, although they are more commonly used to treat acute attacks. Agents that reduce uric acid synthesis or increase the renal excretion of uric acid prevent monosodium urate crystal formation and are useful for prevention of recurrent attacks. These pharmacologic interventions provide effective therapy for most cases of gout.
 Mr. J, a 53-year-old man, awakens one morning with excruciating pain in his great toe. Even the weight of the bed sheet is enough to make him want to scream; he is unable to put on a sock or shoe. Worried that something terrible has occurred, Mr. J rushes to his doctor. Based on the history and physical findings, the physician diagnoses an acute attack of gout. The physician prescribes a course of high-dose ibuprofen, which improves his symptoms in the first day and relieves the pain after 3 days. Mr. J is then well until 5 years later, when the symptoms recur, and he treats himself with ibuprofen successfully. Subsequently, Mr. J learns to anticipate the attacks, which over the next 10 years slowly increase in frequency until they occur once weekly. He uses ibuprofen at the first hint of pain.
Mr. J, a 53-year-old man, awakens one morning with excruciating pain in his great toe. Even the weight of the bed sheet is enough to make him want to scream; he is unable to put on a sock or shoe. Worried that something terrible has occurred, Mr. J rushes to his doctor. Based on the history and physical findings, the physician diagnoses an acute attack of gout. The physician prescribes a course of high-dose ibuprofen, which improves his symptoms in the first day and relieves the pain after 3 days. Mr. J is then well until 5 years later, when the symptoms recur, and he treats himself with ibuprofen successfully. Subsequently, Mr. J learns to anticipate the attacks, which over the next 10 years slowly increase in frequency until they occur once weekly. He uses ibuprofen at the first hint of pain.
The morning after one of his attacks begins, Mr. J goes to his doctor because the pain is not relieved adequately with ibuprofen. Focused examination reveals a swollen, red, and warm left knee, right midfoot, and right first metatarsophalangeal joint. There are 0.5-cm mobile nodules near the olecranon bilaterally and another at the inferior pole of the right patella. The rest of the exam is unremarkable. The physician aspirates Mr. J’s left knee, revealing a cloudy yellow fluid that, on microscopic examination, contains numerous leukocytes. Abundant blue and yellow needle-shaped microscopic crystals are seen with the use of a polarizing filter with a red compensator, some of them intracellular. An x-ray of the left knee is normal except for the presence of an effusion; a film of the right foot shows a bony erosion of the distal first metatarsal. The culture of the joint fluid is negative.
Mr. J is treated with high-dose prednisone on the first day, followed by a tapering dose over the next 10 days. His condition improves rapidly. Three weeks later, he returns to his physician while feeling well. He is given a prescription for allopurinol to take on a long-term basis and one for colchicine to take during the first 6 months of allopurinol therapy.
Questions
1. Why was ibuprofen effective for most of Mr. J’s acute attacks of pain?
2. How does prednisone reduce the inflammatory response during an acute attack of gout?
3. How does allopurinol act? Will it alter the frequency of Mr. J’s painful attacks?
4. Why was Mr. J prescribed colchicine during the first 6 months of treatment with allopurinol?
 PHYSIOLOGY OF PURINE METABOLISM
PHYSIOLOGY OF PURINE METABOLISM
Gout is caused by imbalances in purine metabolism. To understand the cause and treatment of gout, it is necessary to recall the principles of nucleotide biochemistry. Although pyrimidines such as cytosine, thymidine, and uracil are straightforward for the body to metabolize and excrete, it is a challenge to metabolize purines (most notably the nucleotides guanine and adenine). The intermediates of purine metabolism are toxic to some cells, necessitating tight regulation of purine synthesis and degradation. Furthermore, the final breakdown product of purine metabolism is uric acid, which is barely soluble in blood or urine. Increased plasma levels of uric acid are the strongest risk factor for gout, although, for poorly understood reasons, not everyone with high plasma uric acid levels develops gout.
Purines are synthesized via two general pathways: de novo synthesis and the salvage pathway (Fig. 49-1). The first step in the de novo pathway is the reaction of phosphoribosyl pyrophosphate (PRPP, a ribose sugar with two pyrophosphates attached) with glutamine. PRPP provides the ribose sugar as one precursor for the nascent nucleotide. Hydrolysis of the pyrophosphate in a later step makes the de novo pathway irreversible. Glutamine is the precursor for inosine monophosphate (IMP), a precursor that is common to adenine and guanine biosynthesis. The reaction of PRPP with glutamine is catalyzed by the enzyme amidophosphoribosyltransferase (amidoPRT). AmidoPRT is activated allosterically by high levels of PRPP; PRPP is thus both a substrate and an activator of amidoPRT. In general, the cellular level of PRPP is the most important determinant of de novo purine synthesis. High PRPP levels result in enhanced de novo purine synthesis, whereas low PRPP levels decrease the rate of synthesis.
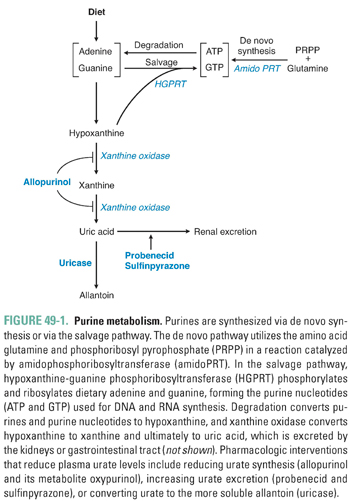
The salvage pathway is the second important route to purine synthesis. The first step in the salvage pathway is catalyzed by the key regulatory enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). HGPRT transfers PRPP to either hypoxanthine or guanine, resulting in the formation of IMP or guanosine monophosphate (GMP), respectively. Nucleotide interconversions can then yield adenosine triphosphate (ATP) and guanosine 5′-triphosphate (GTP).
Increased activity of the salvage pathway has two important consequences. First, increased scavenging activity depletes cells of PRPP, thus decreasing the rate of de novo purine synthesis. Second, the salvage pathway leads to the generation of more ATP and GTP. Increased levels of these nucleotides inhibit amidoPRT in a feedback manner, also resulting in decreased de novo purine synthesis.
Although purines can be synthesized by these two interrelated pathways, degradation occurs via a convergent mechanism (Fig. 49-1). Adenosine monophosphate (AMP) is deaminated, dephosphorylated, and deribosylated, forming hypoxanthine. GMP is also deaminated, dephosphorylated, and deribosylated, forming hypoxanthine. Hypoxanthine, which is moderately soluble, is oxidized to xanthine. Thus, xanthine is the common product of purine metabolism. A further oxidation step converts xanthine to uric acid. The enzyme xanthine oxidase catalyzes both the oxidation of hypoxanthine to xanthine and the oxidation of xanthine to uric acid.
Crosstalk between the de novo and salvage pathways is important for overall regulation of purine metabolism. The de novo pathway is the most important generator of purine breakdown products. High de novo pathway activity increases purine turnover, resulting in higher plasma uric acid concentrations. In contrast, increased salvage pathway activity leads to decreased de novo synthesis and reduced plasma uric acid levels.
The importance of crosstalk in purine metabolism is demonstrated by several inherited enzyme disorders. Certain genetic polymorphisms that increase PRPP synthase activity lead to increased intracellular levels of PRPP; because PRPP activates amidoPRT, high levels of PRPP cause increased de novo purine synthesis, leading to increased turnover and degradation of purines and increased plasma levels of uric acid. Similarly, genetic deficiencies of HGPRT (the critical enzyme in the salvage pathway) lead to decreased salvage pathway activity and increased de novo purine synthesis and degradation, resulting in increased uric acid levels. The inherited absence of HGPRT results in Lesch-Nyhan syndrome, a devastating disorder characterized by self-mutilation, mental retardation, and hyperuricemia. Partial defects in HGPRT (e.g., polymorphisms in the HGPRT gene that lead to decreased HGPRT synthesis or activity) are thought to explain some cases of hereditary gout.
Uric acid is eliminated by the kidney (65%) and gastrointestinal tract (35%). Uric acid is filtered and secreted by the kidney by the same mechanisms that process other organic anions. Approximately 90% of filtered uric acid is reabsorbed. The major mediator of uric acid reabsorption is urate transporter 1 (URAT1), a member of the organic anion transporter family (SLC22A12) that is expressed in the kidney proximal tubule (see Chapter 5, Drug Transporters). Recent genetic association studies have suggested that polymorphisms in URAT1 may predispose to the development of gout. Renal excretion is important for the maintenance of normal plasma uric acid levels, and kidney failure often leads to high plasma urate levels.

The likelihood of developing gout correlates strongly with increased plasma uric acid levels. Uric acid is a weak acid (pKa = 5.6); at physiologic pH, 99% of plasma uric acid is in the ionized, urate form. The normal urate concentration in human plasma is 4–6 mg/dL, reflecting a balance of urate synthesis, breakdown, and excretion. Urate is sparingly soluble: the plasma becomes saturated if urate levels exceed 6.8 mg/dL. A plasma level over 7.0 mg/dL for men or 6.0 mg/dL for women is classified clinically as hyperuricemia. The gender difference may be attributable to differences in urate excretion between women and men.
Any variable that decreases the solubility of urate can promote urate crystal deposition. Gout occurs most commonly in peripheral joints. Urate is less soluble at lower temperatures, which may explain the peripheral distribution of urate crystal deposition. Also, joint synovial fluid is more acidic than blood, favoring crystal formation. Nevertheless, a complete explanation for the pattern of joint involvement in gout remains elusive.
The pathogenesis of gout is thought to reflect deposition of urate crystals in the periarticular fibrous tissue of synovial joints after years of hyperuricemia. However, it is also possible to develop gout without hyperuricemia (e.g., due to an immune response to urate or to preferential deposition of urate in synovial fluid).
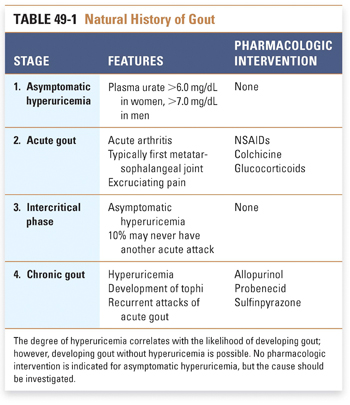
The natural history of gout has four stages (Table 49-1). First, asymptomatic hyperuricemia develops because of either increased purine breakdown or decreased urate excretion. Most cases of hyperuricemia never develop into gout, and there is no indication for treatment of hyperuricemia in the absence of gout. It is, however, important to determine the cause of marked hyperuricemia: such causes can include lymphoma (increased purine turnover) and kidney failure (decreased urate excretion).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


