Innate Responses to Viral Infections
Akiko Iwasaki
Ruslan Medzhitov
The innate immune system provides a universal form of host protection from infectious diseases. It detects the presence of pathogens using several recognition strategies. The best-characterized innate microbial sensing mechanism is based on pattern recognition receptors (PRRs). These receptors detect conserved microbial structures shared by entire classes of microorganisms. The targets of PRRs are commonly products of metabolic pathways unique to a particular class of microbes, such as lipopolysaccharide in the case of Gram-negative bacteria. In the case of viral pathogens, however, all viral molecular constituents are produced in the host cell. Consequently, the major targets of innate immune recognition are viral nucleic acids. Whenever possible, the innate immune system detects structural features of viral RNA and DNA that are distinct from the host nucleic acid. These include long double-stranded RNA, RNAs containing 5′-triphosphate, and unmethylated CpG motifs in viral DNA genomes. Detection of these structural features, however, is insufficient to reliably distinguish the host and viral nucleic acids. Therefore, additional factors help to determine their origin. For example, viral but not host nucleic acids are normally found in the endolysosomes and viral but not host DNA is present in the cytosol. Innate immune sensors of viral RNA and DNA are present in these locations, where they can be triggered to activate the antiviral responses. In addition to pattern recognition, viral pathogens might be detected indirectly, through alteration in normal cellular processes, such as acute decline in host protein synthesis, altered activity of ion channels, or ER stress. These forms of recognition are still incompletely understood and remain to be fully characterized in future studies.
Detection of viral pathogens by the innate immune system has two major consequences: first, it leads to the induction of the innate antiviral mechanisms, most of which are mediated primarily by type-I interferons (IFNs). Second, it leads to the activation of the adaptive immune response that can provide a more directed, antigen-specific, and long-lasting antiviral immunity. The main host defense strategy against viral pathogens is the elimination of the infected cells. This can be achieved by cell-intrinsic mechanisms that are induced by type-I IFNs and operate in the infected cells, or with the help of cytotoxic lymphocytes: natural killer (NK) cells and CD8 T cells. Another important host defense mechanism is the blockade of viral entry into the host cells. This is primarily a function of neutralizing antibodies. Many additional mechanisms exist that interfere with viral replication, gene expression, virion assembly, and exit from the infected cells. The relative contribution of these defense mechanisms varies depending on the virus and host. Detailed knowledge of the specific antiviral defense mechanisms is difficult to establish due in large part to a high degree of redundancy between different mechanisms.
In this chapter, we will focus on the better-characterized viral sensing mechanisms of the innate immune system and discuss the major defense mechanisms utilized in mammalian hosts.
In this chapter, we will focus on the better-characterized viral sensing mechanisms of the innate immune system and discuss the major defense mechanisms utilized in mammalian hosts.
Sensing Viral Infections
Viral Sensing Through Pattern Recognition Receptors
The mammalian immune system detects the presence of viruses through multiple mechanisms. The best understood mechanisms involve various PRRs. Most common molecular patterns associated with virus infection are the features associated with viral nucleic acids. Innate immune recognition can be cell intrinsic or cell extrinsic, depending on whether it is mediated by infected or non-infected cells. Cell-intrinsic innate immune recognition is mediated by cytosolic sensors, including the NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs). Activation of these receptors generally occurs in infected cells. Accordingly, PRRs involved in cell-intrinsic recognition are broadly expressed because viral pathogens target a variety of cell types for replication. In contrast, cell-extrinsic innate immune recognition is mediated by transmembrane receptors (including Toll-like receptors (TLRs) and C-type lectins [CLRs]); their activation does not require the cells expressing these receptors to be infected. Cell-extrinsic recognition is mainly mediated by specialized cells of the immune system, such as the plasmacytoid dendritic cell (pDC), macrophage, and dendritic cell (DC).
PRRs involved in microbial recognition have modular structures and share several structurally related domains for innate recognition, multimerization, interaction with adaptor molecules, and signaling (Fig. 8.1).
Cell-Autonomous Virus Recognition
RIG-I-Like Receptors (RLRs)
RNA viruses are recognized by the cytoplasmic RNA helicases, retinoic acid–inducible gene-I (RIG-I), and melanoma differentiation–associated gene 5 (MDA5).198 The RIG-I-like receptors (RLRs) are expressed by most cell types, and both RIG-I and MDA5 recognize viral RNA through their helicase domains. RIG-I also contains the repressor domain (RD) at its C-terminus, which inhibits the activation of RIG-I at steady state. Binding to viral RNA catalyzes a conformational rearrangement, which exposes a caspase activation and recruitment domain (CARD) to initiate antiviral signaling152,199 (Fig. 8.1). Both RIG-I and MDA5 utilize a common adaptor molecule called mitochondria antiviral signaling protein (MAVS)162—also known as IPS-1,93 Cardif,127 or VISA194—that localizes to the mitochondrial membrane162 and to the peroxisomes.47 MAVS activates two protein kinase complexes, one consisting of FADD and caspases 8/10175 and the other containing TANK and NAP1155 (Fig. 8.2). The former activates the IKKα/IKKβ/IKKγ complex while the latter activates the TBK1-IKKi complex, leading to the activation and nuclear translocation of transcription factors NF-κB and IRF3, respectively. In addition, the TRAF6-dependent pathway involving MEKK1 is essential to activate the MAP kinase and NF-κB pathways.201 Despite similarity in overall structure and signaling mechanisms, RIG-I and MDA-5 have important differences in their mechanisms of activation. For example, RIG-I but not MDA5 requires ubiquitination by E3 ligase TRIM25 for its activity.58 Additional differences in the function of RIG-I and MDA-5 likely exist and may reflect distinct features of viral pathogens that they recognize.
Analysis of animals deficient in the RLRs revealed important and distinct roles for these sensors in innate immunity. These molecules cumulatively provide the host with the ability to recognize a large group of viral pathogens in infected cells and mediate a critical aspect of innate antiviral defense.
RIG-I
RIG-I consists of two N-terminal CARDs, a central DExH box RNA helicase/adenosine triphosphatase (ATPase) domain, and a C-terminal regulatory domain (RD) (Fig. 8.1). RIG-I functions as a critical PRR for a number of viruses including Sendai virus (SeV), vesicular stomatitis virus (VSV), influenza, hepatitis C virus, Japanese encephalitis virus, as well as small RNA encoded by the DNA virus, Epstein-Barr virus.92 RIG-I
recognizes single-stranded (ssRNA) that contains 5′ triphosphate end, but not 5′ OH or a 5′-methylguanosine cap73,144 that is longer than 23 nucleotides123 and contains a uridine- or adenosine-rich ribonucleotide sequence153 (Fig. 8.2). RIG-I can also recognize double-stranded (dsRNA) in the absence of 5′ triphosphate in some cases with a preference for shorter-length dsRNA compared to those recognized by MDA-5.91 Analysis of the central DExH box RNA helicase/ATPase domain of RIG-I revealed an ATP-powered dsRNA translocation activity. The CARD domains suppress translocation in the absence of 5′-triphosphate, and the activation by 5′-triphosphate triggers RIG-I to translocate preferentially on dsRNA in cis.130 This ATPase activity is required for RIG-I signaling, indicating that RIG-I recognizes two distinct features of viral RNA simultaneously: triphosphate at the 5′ end and dsRNA. RIG-I indeed selectively detects blunt, short, double-stranded 5′-triphosphate RNA, which is common in the panhandle region of ssRNA viral genomes.157
recognizes single-stranded (ssRNA) that contains 5′ triphosphate end, but not 5′ OH or a 5′-methylguanosine cap73,144 that is longer than 23 nucleotides123 and contains a uridine- or adenosine-rich ribonucleotide sequence153 (Fig. 8.2). RIG-I can also recognize double-stranded (dsRNA) in the absence of 5′ triphosphate in some cases with a preference for shorter-length dsRNA compared to those recognized by MDA-5.91 Analysis of the central DExH box RNA helicase/ATPase domain of RIG-I revealed an ATP-powered dsRNA translocation activity. The CARD domains suppress translocation in the absence of 5′-triphosphate, and the activation by 5′-triphosphate triggers RIG-I to translocate preferentially on dsRNA in cis.130 This ATPase activity is required for RIG-I signaling, indicating that RIG-I recognizes two distinct features of viral RNA simultaneously: triphosphate at the 5′ end and dsRNA. RIG-I indeed selectively detects blunt, short, double-stranded 5′-triphosphate RNA, which is common in the panhandle region of ssRNA viral genomes.157
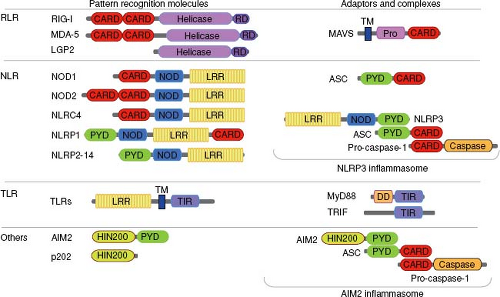 Figure 8.1. Domain structure of viral sensors and adaptor molecules. Pattern recognition molecules involved in viral sensing contain distinct domains, some of which are shared by different classes of receptors. These domains include: CARD, caspase activation and recruitment domain; DD, death domain; HIN200, hemopoietic IFN-inducible nuclear proteins is a 200-amino acid motif; LRR, leucine-rich repeat; NOD, nucleotide-binding oligomerization domain; Pro, proline-rich region; PYD, pyrin domain; caspase domain; TIR, toll/interleukin-1 receptor; TM, transmembrane region. Many of these domains can be used for homotypic oligomerization. Examples of molecular complexes are depicted for NLRP3 and AIM2 inflammasomes. |
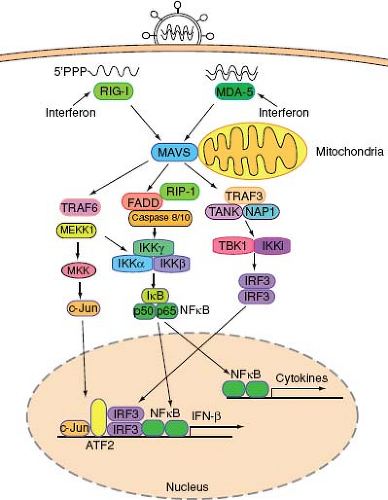 Figure 8.2. RIG-I and MDA-5 virus recognition pathways. In a virally infected cell, RNA structures that are unique to viruses are recognized by RIG-I and MDA-5. RIG-I detects ssRNA containing 5′ triphosphate, while MDA-5 recognizes a long stretch of dsRNA. RNA binding to RLRs induces conformational changes, enabling their binding to MAVS adaptor protein expressed on the surface of mitochondria and peroxisomes. MAVS activates signaling to activate NF-κB, MAP kinases, and IRF3, leading to target gene expression including IFN-β and cytokines. Type I IFNs increase the levels of RLRs in bystander cells, enabling robust antiviral signaling upon infection. |
MDA5
MDA5 serves as a sensor of picornaviruses and can be activated by cytosolic synthetic dsRNA, such as polyinosinic-polycytidylic acid (poly I:C).60,92 There appears to be some level of redundancy in RIG-I and MDA5 in recognition of certain viruses. Dengue virus and West Nile virus are recognized by both MDA5 and RIG-I; either of these sensors is sufficient to induce type I IFN production.115 Comparison of synthetic and viral dsRNA revealed that MDA-5 preferentially recognizes longer dsRNA that is at least 2 k bp in length, while RIG-I is activated by a wider range of dsRNA sizes (400 bp–4 kbp).91 Many viruses that replicate in the cytoplasm express their own 2′-O-methyltransferases to autonomously modify their messenger RNA (mRNA). Interestingly, infection with viruses deficient in 2′-O-methyltransferase leads to robust activation of MDA5, indicating that lack of ribose 2′-O-methylation is used as a signature of viral RNA for recognition by MDA5.205 Therefore, MDA5 likely recognizes large dsRNA structures lacking 2′-O-methylation generated in the cytosol during virus infection. Of note, 2′-O-methylation is also used by viruses to evade restriction by IFN-induced proteins with tetratricopeptide repeats (IFIT) proteins.36,205
LGP2
The third member of the RLR family, laboratory of genetics and physiology 2 (LGP2), displays sequence homology to the helicase domains of RIG-I and MDA5. It lacks a CARD domain, however, and therefore has been proposed to serve as a negative regulator of both sensors. Overexpression studies indicated that LGP2 does not activate the production of type I IFNs on its own, but inhibits RIG-I- and MDA5-mediated signaling in a dose-dependent manner.98,149,200 Interestingly, studies of lgp2 knockout mice indicated that LGP2 may play both positive and negative regulatory roles in MDA5- and RIG-I-mediated innate immunity.189 This study found that the loss of lgp2 augments type I IFN production in response to transfected poly I:C (MDA-5 agonist) or VSV infection (RIG-I agonist). In contrast, LGP2 is required for efficient antiviral responses following EMCV infection (MDA5 agonist). Therefore, LPG2 can serve as a positive or negative regulator of RLR signaling depending on the type of viral infection. The precise mechanism underlying the opposing functions of LPG2 is still unclear.
NOD-Like Receptors (NLRs)
NLRs comprise a large family of intracellular PRRs that regulates innate immunity in response to recognition of various PAMPs and stress signals.125 The NLR family consists of multidomain proteins that contain a C-terminal LRR domain, a central NOD domain and an N-terminal effector domain. NLR proteins can be subdivided into three subfamilies depending on the structure of N-terminal domains: CARD-containing subfamily (NOD1, NOD2, NLRCs, CIITA), pyrin domain (PYD)–containing subfamily (NLRPs) (Fig. 8.1), and BIR domain–containing subgroup (NAIPs) (not shown). Both NOD and NLRP members contribute to antiviral defense. The PYD subfamily of NLRPs consists of 14 members. Although the functions of many of the NLRPs are largely unknown, several NLRPs play a key role in the activation of caspase-1 by forming a multiprotein complex known as the
“inflammasome”124 (Fig. 8.3). Caspase-1 is an essential mediator of inflammatory response through its capacity to cleave and generate active forms of IL-1β and IL-18. IL-1β and IL-18 are potent proinflammatory cytokines, as described below. Formation and secretion of mature IL-1β and IL-18 require a two-step activation mechanism: first, transcriptional and translational upregulation of the pro-forms of these cytokines are induced by TLR signaling, and a second signal that leads to the proteolytic activation of caspase-1. The latter process is mediated by the inflammasome. Inflammasome complexes that are important in antiviral defense are described below.
“inflammasome”124 (Fig. 8.3). Caspase-1 is an essential mediator of inflammatory response through its capacity to cleave and generate active forms of IL-1β and IL-18. IL-1β and IL-18 are potent proinflammatory cytokines, as described below. Formation and secretion of mature IL-1β and IL-18 require a two-step activation mechanism: first, transcriptional and translational upregulation of the pro-forms of these cytokines are induced by TLR signaling, and a second signal that leads to the proteolytic activation of caspase-1. The latter process is mediated by the inflammasome. Inflammasome complexes that are important in antiviral defense are described below.
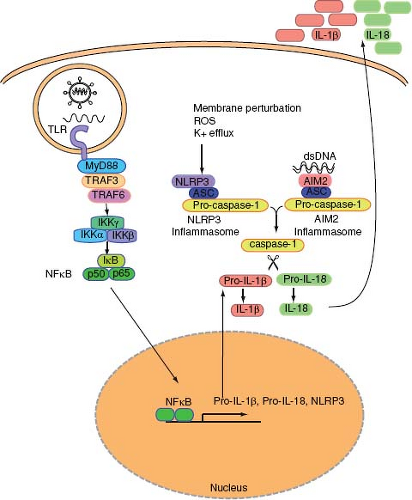 Figure 8.3. Inflammasome activation by virus infection. Virus infection can lead to inflammasome activation through two separate pathways. NLRP3 inflammasome is activated upon infection by adenovirus or influenza virus. This pathway is initiated by membrane perturbation, K+ efflux, and reactive oxygen species (ROS). The second pathway involves AIM2, which is activated by dsDNA in the cytosol. Both NLRP3 and AIM2 form a complex with ASC and pro-caspase-1 (inflammasome), leading to the self-cleavage and activation of caspase-1. Pro-forms of IL-1β and IL-18 induced by TLR signals are cleaved by activated caspase-1. Mature forms of these cytokines are then released to the extracellular space. |
NLRP3-ASC Inflammasome
NLRP3, also known as NALP3/Cryopyrin/CIAS1/PYPAF1,181 forms an ASC-dependent inflammasome (Fig. 8.3). The NLRP3 inflammasome can be activated by a variety of stimuli including endogenous signals from dying cells (uric acid), crystals (asbestos, silica, alum), as well as microbial signals such as whole bacteria, bacterial RNA, extracellular ATP, pore-forming toxins, or viral infections.125 It is unclear whether microbial ligands can directly activate the NLRP3 inflammasome. Instead, the NLRP3 inflammasomes likely sense cellular stress such as disruption in membrane integrity and extracellular ATP released from stressed or damaged cells. Virus infection also results in the activation of inflammasomes. Both SeV and influenza virus activated the NLRP3 inflammasome in macrophages pulsed transiently with ATP (signal 2) in vitro.90 DNA viruses such as adenovirus stimulate the NLRP3-ASC-caspase-1 inflammasomes in vivo.129 However, inflammasomes are not activated by transfection of RNA, poly I:C, or infection with reovirus (dsRNA virus) or VSV (ssRNA virus),129 indicating that viral RNAs are insufficient to trigger inflammasome activation. Influenza virus activates the inflammasome through the activity of the M2 ion channel.79 The M2 channel of influenza A virus is a homotetrameric integral membrane protein that associates to form a highly specific proton channel,145 and is essential for influenza A virus infection and replication.179 Among other things, the M2 channel exports protons in the acidic trans-Golgi network (TGN) to neutralize the pH of the lumen of the TGN in order to prevent the premature maturation of hemagglutinin to its low-pH fusogenic form.32 Within influenza virus–infected cells, NLRP3 complex detects changes in ionic imbalance in the TGN to activate the inflammasomes. In vivo, NLRP3 deficiency resulted in increased susceptibility to high-dose flu challenge.4,180 In addition, after a sublethal dose of influenza infection, ASC-dependent inflammasome activation is required to elicit adaptive protective immunity to influenza virus, indicating the importance of NLRs in linking innate viral recognition to adaptive immunity.78
AIM2-ASC Inflammasome
Not all inflammasomes are activated by NLRPs. Recent studies showed that AIM2 couples cytosolic dsDNA recognition to ASC-caspase-1 inflammasome.25,53,72,148 AIM2 is an HIN200 family of protein that contains a dsDNA binding domain (HIN domain) and the PYD domain, which promotes interaction with the PYD domain of ASC (Fig. 8.1). AIM2 recognizes dsDNA in the cytosol and induces oligomerization of ASC and caspase-1, leading to activation of caspase-1 and cleavage of pro-forms of cytokines including IL-1β, IL-18, and IL-33 (Fig. 8.3). Vaccinia virus, which contains a dsDNA genome, was shown to require AIM2 for recognition and activation of caspase1 inflammasomes.72 It is interesting to note that different classes of dsDNA viruses are recognized by NLRP3 (adenovirus) or AIM2 (vaccinia virus) for inflammasome activation. This likely depends on both the accessibility of the dsDNA genome to intracellular sensors and the viral-induced cellular stress responses. While cytosolic dsDNA can bind to AIM2 directly and activate inflammasomes, how and where exactly NLRP3 becomes activated by dsDNA viruses is yet to be determined.
p202
Mouse HIN200 protein, p202, is a negative regulator of the AIM2 inflammasome.148 No orthologs of p202 have been found in humans. p202 contains two HIN domains but no PYD domain, and binds specifically to dsDNA. Unlike AIM2, p202 cannot bind ASC and therefore appears to function as an inhibitor of AIM2, presumably by limiting its access to DNA.
Cytosolic DNA Sensors
The existence of a cytoplasmic DNA sensing molecule leading to type I IFN production was suggested from studies of
infection by DNA viruses and bacteria.82,170 It is now clear that there are likely multiple sensors that recognize distinct types of DNA. These DNAs include DNA from viruses, bacteria, apoptotic host cells, and synthetic B-form DNAs—particularly poly(dA:dT) and interferon stimulatory DNA (ISD). ISDs are dsDNA containing >25 base-pair oligonucleotides, which in sequence-independent manner, trigger stimulation of type I IFNs through a pathway involving TBK1 and IRF3, but not MAVS.170 Interestingly, ISD does not engage NK-κB or MAPK pathways, thus activating only IRF3-induced pathways. ISD recognition pathway exists only in primary cells but is lost from transformed cells.170 However, the sensor for ISD still remains to be identified. TREX1, an exonuclease, was identified to be a negative regulator of ISD pathway by inhibiting excess accumulation of DNA products from endogenous retroelements.169 TREX1-deficient humans and mice suffer from autoimmune disease due to hyperstimulation of ISD pathway. TREX1 is also involved in degrading nonproductive RT products generated during HIV-1 infection, enabling HIV-1 to remain undetected by the ISD sensor.195
infection by DNA viruses and bacteria.82,170 It is now clear that there are likely multiple sensors that recognize distinct types of DNA. These DNAs include DNA from viruses, bacteria, apoptotic host cells, and synthetic B-form DNAs—particularly poly(dA:dT) and interferon stimulatory DNA (ISD). ISDs are dsDNA containing >25 base-pair oligonucleotides, which in sequence-independent manner, trigger stimulation of type I IFNs through a pathway involving TBK1 and IRF3, but not MAVS.170 Interestingly, ISD does not engage NK-κB or MAPK pathways, thus activating only IRF3-induced pathways. ISD recognition pathway exists only in primary cells but is lost from transformed cells.170 However, the sensor for ISD still remains to be identified. TREX1, an exonuclease, was identified to be a negative regulator of ISD pathway by inhibiting excess accumulation of DNA products from endogenous retroelements.169 TREX1-deficient humans and mice suffer from autoimmune disease due to hyperstimulation of ISD pathway. TREX1 is also involved in degrading nonproductive RT products generated during HIV-1 infection, enabling HIV-1 to remain undetected by the ISD sensor.195
In contrast, B-form DNA—or poly(dA-dT)·poly(dT-dA) dsDNA—triggers type I IFNs via IRF3 and NK-κB, suggesting that this form of DNA triggers a separate sensor from ISD.81 The sensor(s) for cytosolic DNA remained elusive until recently. First, a molecule called DAI (also known as DLM-1/ZBP1) was identified as a candidate intracellular DNA sensing molecule.176 IFN induction following poly(dA-dT)·poly(dT-dA) DNA treatment was abrogated by a small interfering RNA (siRNA) directed against DAI, suggesting that DAI is a critical cytoplasmic DNA sensor.176 However, DAI is not the sole sensor of DNA in the cytosol, as DAI-knockout mice still responded to B-DNA or plasmid DNA.83 More recent studies showed that poly(dA-dT)·poly(dT-dA) DNA is recognized by RIG-I upon transcription by RNA polymerase III (Pol III).1,30 Pol III synthesizes 5′-triphosphate RNA using AT-rich dsDNA as a template, generating an RIG-I agonist in the cytosol. Therefore, RIG-I is the sensor for cytosolic poly(dA-dT)·poly(dT-dA) dsDNA upon transcription by Pol III. In addition to the indirect sensing of poly(dA-dT)·poly(dT-dA) by RIG-I, IFI16, a PYHIN protein, can bind to poly(dA-dT)·poly(dT-dA), and knockdown of IFI16 expression led to reduction in IFN-β production.186
Cyclophilin A
While most pattern recognition of virus infection involves detection of nucleic acids, there are few exceptions in which viral protein serves as a signature for viral infection. Cyclophilin A is a peptidylprolyl isomerase that can catalyze cis/trans isomerization of X-Proline epitopes on target proteins. Innate response to HIV-1 in DCs depends on interaction between the newly synthesized HIV-1 capsid and cellular cyclophilin A, leading to activation of type I IFNs through IRF-3-dependent mechanisms.122 Human DCs, but not CD4 T cells, have intrinsic machinery for responding to HIV-1 infection through cyclophilin A. However, infected humans are unable to utilize the cyclophilin A–dependent pathway of IFN induction against HIV-1 infection because DCs are resistant to HIV-1 infection. In addition, cyclophilin A is also known to be incorporated into the HIV-1 virions by binding to capsid protein. Virions lacking cyclophilin A possess normal morphology and can penetrate host cells, but are defective in the reverse transcription of viral RNA.22 Therefore, HIV-1 utilizes cyclophilin A for its infectivity, and renders cyclophilin A useless in antiviral defense by avoiding infection in the relevant cell type.
Cell-Extrinsic Virus Recognition
Toll-Like Receptors (TLRs)
TLRs are the best characterized members of the PRR family, which recognize evolutionarily conserved molecular patterns associated with a large variety of microorganisms.87 There are 11 functional TLRs in mice (TLR1–7, 9, 11–13) and 10 functional TLRs in humans (TLR1–10). All TLRs are type I transmembrane proteins with ectodomains containing leucine rich repeat (LRR) that mediate the recognition of PAMPs. Some TLRs recognize bacterial, fungal, and protozoan pathogens, while others are dedicated for viral recognition.177 This chapter will focus on the TLRs that recognize viral pathogens.
The transmembrane domains of TLRs dictate the localization of the receptor, and the cytosolic domain contains the toll/interleukin-1 receptor (TIR) domain, which binds to adaptor proteins and ultimately leads to expression of a variety of genes.2 TLR activation, following virus detection, catalyzes a complex signaling cascade that bifurcates into two main pathways, culminating in the synthesis of proinflammatory cytokines (NF-κB-dependent) and antiviral cytokines, type I interferons (IFNs) (IRF-dependent). In addition, TLR signaling results in the activation of the mitogen-activated protein kinases (MAPKs) (see Fig. 8.5). Type I IFNs in turn induce a battery of genes that directly suppress viral replication. Other cytokines and chemokines efficiently recruit immune cells to sites of virus infection. In particular, TLR recognition by DCs, the most potent antigen-presenting cells, enables their activation and antigen-presenting function, effectively linking innate recognition to the induction of adaptive immunity.86
TLRs are distributed on specific subcellular locations and are specifically expressed by different cell types. TLRs can be categorized into two major types: those that are expressed on the cell surface (TLR1, 2, 4, 5, 6, 11, and 12) and those that are expressed in the endolysosomes (TLR3, 7, 8, and 9). Although virus–TLR interaction can occur with both types of TLRs, many of the virus interactions with surface TLRs represent viral invasion mechanism (beneficial to the virus) rather than PAMP recognition by TLRs (beneficial to the host) (see discussion under Virus Manipulation of Innate Immunity). In this section, we will discuss the latter type of TLR-virus PAMP recognition leading to antiviral defense.
Viruses are recognized by a group of TLRs that reside in the endosomal membrane—namely, TLR3, 7, 8, and 9—where they can gain access to viral nucleic acids upon endocytosis of virions. A major advantage of this strategy of viral recognition is that the viral sensing and subsequent induction of type I IFN genes are not subjected to viral evasion mechanisms. In addition, since all viruses contain genomes consisting of RNA or DNA, such molecular patterns can be surveyed by a very limited number of receptors. These endosomal TLRs require trafficking by the endoplasmic reticulum (ER) membrane protein, UNC-93B.24,174 UNC-93B physically interacts with TLR3, 7, and 9 via the transmembrane domain,24 and transports these TLRs from the ER to the endosome (Fig. 8.4). Mutations in the Unc93b1 gene were found in two patients with severe HSV encephalitis,26 indicating the importance of transport of endosomal TLRs via
UNC93B in antiviral defense. Upon arrival in the endosomal compartment, these TLRs undergo processing by endosomal proteases. While full-length and cleaved forms of TLR9 are capable of binding ligands, only the processed form recruits MyD88 upon activation.52,140 The ectodomain of TLR9, and likely TLR3 and TLR7,52 must first be cleaved in the endolysosomes by cathepsins in order to become competent for signaling (Fig. 8.4). Dependence of TLR9 activation on proteolytic processing in the endolysosomes ensures that aberrant activation of TLR9 from cell surface (for example, upon recognition of self DNA) is avoided.
UNC93B in antiviral defense. Upon arrival in the endosomal compartment, these TLRs undergo processing by endosomal proteases. While full-length and cleaved forms of TLR9 are capable of binding ligands, only the processed form recruits MyD88 upon activation.52,140 The ectodomain of TLR9, and likely TLR3 and TLR7,52 must first be cleaved in the endolysosomes by cathepsins in order to become competent for signaling (Fig. 8.4). Dependence of TLR9 activation on proteolytic processing in the endolysosomes ensures that aberrant activation of TLR9 from cell surface (for example, upon recognition of self DNA) is avoided.
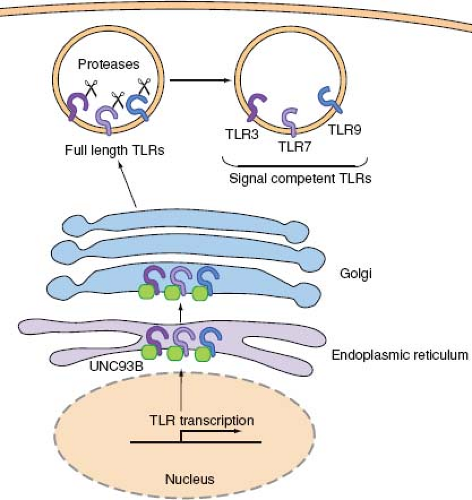 Figure 8.4. Endosomal TLR trafficking and processing. Endosomal TLRs, TLR3, TLR7, and TLR9, are synthesized in the ER, transported through Golgi, and are sorted into endosomes. UNC93B is an ER-resident protein, which facilitates TLR transport to the endolysosomes. Once in the endosomes, TLR3, 7, and 9 undergo proteolytic cleavage by proteases, which makes these receptors competent for signaling. |
TLR3
TLR3 is expressed by a variety of cells in human and mice, including conventional DCs, B cells, fibroblasts, and epithelial cells but not in plasmacytoid DCs (pDCs). Upon PAMP recognition, TLR3 recruits TRIF via its TIR domain (Fig. 8.5). TRIF is responsible for activation of both NF-κB and IRF3, leading to proinflammatory cytokines and type I IFN gene expression, respectively. TRIF binds to TRAF6, which is a RING-domain E3 ubiquitin ligase capable of polyubiquitinylating TRAF6 and IKKγ. Ubiquitinylated TRAF6 and IKKγ recruit TAK1 complex (consisting of TAK1 and TAB proteins), leading to the activation of MAPK. TRIF also binds to receptor-interacting protein (RIP)1 to initiate NF-κB activation. To activate the type I IFN genes, TRIF also recruits TRAF3, which engages TBK1 and subsequent activation of IRF3. IRF-3 translocates to the nucleus and binds to promoters of type I IFN genes.
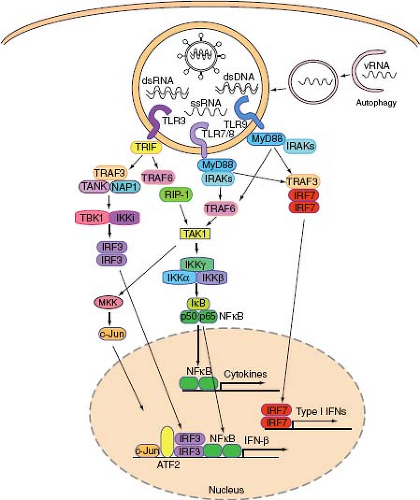 Figure 8.5. Endosomal TLR virus recognition and signaling. Viruses or virus-infected cells are endocytosed, and signatures of virus infection become accessible to TLRs upon digestion of membranes and nucleocapsid. TLR3 recognizes dsRNA, TLR7 and TLR8 recognize ssRNA, and TLR9 recognizes DNA. In addition, autophagy delivers TLR7 ligand (cytosolic viral replication intermediate) by fusion with the endosomes containing TLR7. Proteolytically activated TLRs in the endosomes recruit adaptor proteins and induce downstream signaling to activate a variety of transcription factors. IRF3 in combination with NF-κB, c-Jun, and ATF bind to the promoter and induce IFN-β expression. IRF7 homodimer binds to promoter regions of IFN-a genes and induces IFN-a expression. In addition, NF-κB activates transcription of a large number of genes, including cytokines. |
dsRNA intermediates are produced during the replication cycle of most RNA viruses (except for retroviruses). DNA viruses also produce dsRNA by convergent transcription of their genomes.193 It has long been known that dsRNA triggers IFN production. The first TLR implicated in viral nucleic acid recognition was TLR3.3 TLR3 responds to the artificial dsRNA mimic, poly I:C, when it is provided extracellularly.60,92 However, evidence supporting TLR3-mediated recognition of authentic viral replication intermediates remains elusive, possibly due to overlapping recognition pathways that can compensate for the loss of TLR3. TLR3 may be involved in recognizing virus-produced dsRNA in the context of phagocytized apoptotic cells.159 Humans with dominant negative mutations in the tlr3 gene suffer from neonatal herpes infection,202 indicating the importance of this receptor in protection against HSV-1 in the CNS.
TLR7/8
In humans, TLR7 is expressed in plasmacytoid DCs and B cells, while TLR8 is expressed by myeloid DCs and monocytes. In
mice, TLR7 is expressed by pDCs, conventional DCs (except for CD8α+ DCs), and B cells. The function of the mouse TLR8 is still poorly understood. However TLR8-deficient mice develop autoimmunity due to hyperstimulation of TLR7.39 TLR7, 8, and 9 are all expressed in the endosomes and share similar signaling pathways. Upon engagement, these receptors recruit MyD88 via the TIR domain. MyD88 subsequently interacts with the death domain of several IRAK proteins to induce both NF-κB and IRF7 activation. IRAK4 is required for activation of NF-κB pathway by interacting with TRAF6, which in turn activates the TAK1 complex, leading to activation of NF-κB and MAPK pathways (Fig. 8.5). NF-κB activation follows the recruitment of IRF5 to the MyD88 complex. On the other hand, MyD88 also recruits IRF7, which forms a signaling complex with IRAK4 and TRAF6. TRAF3 also binds MyD88 and IRAK1 to induce IRF7 activation. Unlike TLR3, TLR7 and 9 utilize IRF7 and not IRF3 for activation of type I IFN genes in pDCs. Interestingly, in conventional DCs, TLR7 and 9 utilize MyD88 and IRF1 to induce IFN-β but not IFN-α genes.143
mice, TLR7 is expressed by pDCs, conventional DCs (except for CD8α+ DCs), and B cells. The function of the mouse TLR8 is still poorly understood. However TLR8-deficient mice develop autoimmunity due to hyperstimulation of TLR7.39 TLR7, 8, and 9 are all expressed in the endosomes and share similar signaling pathways. Upon engagement, these receptors recruit MyD88 via the TIR domain. MyD88 subsequently interacts with the death domain of several IRAK proteins to induce both NF-κB and IRF7 activation. IRAK4 is required for activation of NF-κB pathway by interacting with TRAF6, which in turn activates the TAK1 complex, leading to activation of NF-κB and MAPK pathways (Fig. 8.5). NF-κB activation follows the recruitment of IRF5 to the MyD88 complex. On the other hand, MyD88 also recruits IRF7, which forms a signaling complex with IRAK4 and TRAF6. TRAF3 also binds MyD88 and IRAK1 to induce IRF7 activation. Unlike TLR3, TLR7 and 9 utilize IRF7 and not IRF3 for activation of type I IFN genes in pDCs. Interestingly, in conventional DCs, TLR7 and 9 utilize MyD88 and IRF1 to induce IFN-β but not IFN-α genes.143
TLR7 and 8 recognize ssRNA and induce innate immune responses to ssRNA viruses. TLR7 is required for type I IFN and cytokine responses to influenza, SeV, and VSV.44,117 Uridine and ribose, the defining signatures of RNA, are both necessary and sufficient for TLR7 stimulation.45 Furthermore, viral fusion and/or uncoating and endosomal acidification are required for TLR7-dependent recognition.117 Viral RNA, synthetic poly U RNA, and even nonviral, cellular RNA in the endosome is sufficient to stimulate TLR7-dependent cytokine production,44,68 indicating that any RNA localized to the endosome is able to trigger TLR7 activation. While influenza virus is recognized upon endocytosis by TLR7, other viruses such as VSV and SeV require replication in the cytosol prior to recondition by TLR7. The latter type of viruses are recognized by TLR7 in the endosome upon delivery of cytosolic viral replication intermediates through the process of autophagy106 (see below).
TLR9
TLR9 is expressed in pDCs and B cells in humans. Mice express TLR9 in pDCs, B cells, macrophages, and conventional DCs. TLR9 is located in the endosome and mediates recognition of viral DNA. Originally, bacterial DNA sequences containing hypomethylated CpG motifs were shown to activate TLR9.69 More recent studies showed that TLR9 might recognize the sugar-base-backbone, 2-deoxyribose, of phosphodiester DNA irrespective of the CpG content.61 TLR9 is the principal means by which HSV-1 and HSV-2 stimulate type I IFNs in pDCs in vivo.100,116 Interestingly, a TLR9 molecule “retargeted” to the plasma membrane, unable to respond to viral nucleic acids, however now responded to self DNA that did not stimulate wild-type TLR9.15 These results suggest that not only is endosomal localization important to trigger TLR-viral nucleic acid interactions, but that endosomal TLRs may limit access to nonviral nucleic acids via an active sequestering mechanism in the endosome.
C-Type Lectins
In addition to TLRs, C-type lectin receptors (CLRs) expressed on the plasma membrane can bind to certain viruses. Classical CLRs contain carbohydrate recognition domain responsible for the Ca2+-dependent binding to their ligands. However, carbohydrate-recognition domains of many CLRs do not bind to Ca2+.8 CLRs can be classified into those that are used solely for endocytosis of pathogens and those that are used for inducing signaling. The endocytic CLRs include mannose receptor DEC205 and Langerin, which are expressed on CD8α+ DCs and Langerhans cells, respectively. Langerin on Langerhans cells are capable of clearing HIV by receptor-mediated endocytosis and degradation in Birbeck granules.37
The signaling CLRs include Dectin-1 and DC-SIGN.40 Dectin-1, which is a receptor for fungal β-glucans, contains an ITAM motif in the cytosolic domain, and engages Syk tyrosine kinase and CARD9, leading to NF-kB activation.95 Dectin-1 plays an important role in antifungal defense, but it is not known to have antiviral functions. DC-SIGN binds to HIV-1 and Ebola virus. Signaling through DC-SIGN alone does not lead to the activation of NF-κB or expression of cytokines, but can modulate signaling through other PRRs. DC-SIGN engagement by viruses leads to serine/threonine kinase Raf-1 activation. After translocation of NF-κB by TLR-stimulation, DC-SIGN-activated Raf-1 mediates the phosphorylation of NF-κB subunit p65, which in turn leads to p65-acetylation. Acetylation of p65 both prolongs and increases IL-10 transcription, resulting in increased IL-10 production. However, the majority of C-type lectin–virus interactions reflect a viral invasion mechanism. A clear example of this is DC-SIGN signaling by HIV-1. This interaction results in impaired DC maturation, enhanced T-cell proliferation and transmission of HIV-1 to T cells, thereby promoting systemic infection of the host. DC-SIGN and a related C-type lectin, DC-SIGNR, also enhance infection by Ebola virus.13 Another example is Dengue virus, which binds to CLEC5A and induces signaling through DAP12 to induce proinflammatory cytokines. This leads to lethality, not protection, in mice.29
Other Strategies of Viral Recognition
In addition to PRR-based viral recognition, non-PRR-based, cell-autonomous virus sensing mechanisms exist in mammalian hosts. While molecular mechanisms for such pathways are not yet understood, these pathways likely involve sensing various forms of cellular stresses inflicted by virus infection. One of the hallmarks of virus infection is the production of large amounts of viral proteins. This often leads to ER overload, leading to ER stress and unfolded protein response (UPR) induction. Virus infection also often leads to the generation of reactive oxygen species (ROS) by interfering with proper mitochondrial function. In addition, as discussed below, most viruses express molecules that inhibit vital cellular functions such as transcription, translation and secretion. These types of stress that accompany virus infections might be seen by the mammalian host as a signature of virus infection. In conjunction with pattern recognition, such stress sensing pathways can engage signals leading to type I IFNs synthesis, cell-cycle arrest or cell death to avoid further viral replication and spread.
Innate Antiviral Cytokines
Type I Interferons
Antiviral mechanisms in vertebrates are highly dependent on the action of type I IFNs. Type I IFNs are a family of cytokines that act early in the innate immune response and are key
cytokines capable of inducing an antiviral state in infected and uninfected neighboring cells.80 In addition to this antiviral activity, the interferon cytokines have a role in regulating the ensuing adaptive immune response.171 While type II and type III IFNs also have antiviral activities, in this chapter we will focus on the role of type I IFNs in innate antiviral defense. In humans, the type I IFNs consist of 13 α-genes coding for 12 IFN-α subtypes, one β-gene encoding a single IFN-β subtype and a single gene encoding IFN-ω. Type I IFNs bind to IFN-αβR, which is a heterodimer of IFN-αR1 and IFN-αR2 chains. High levels of IFN-α subtypes are rapidly secreted from pDCs upon viral recognition, whereas IFN-β can be induced from most virus-infected cell types.
cytokines capable of inducing an antiviral state in infected and uninfected neighboring cells.80 In addition to this antiviral activity, the interferon cytokines have a role in regulating the ensuing adaptive immune response.171 While type II and type III IFNs also have antiviral activities, in this chapter we will focus on the role of type I IFNs in innate antiviral defense. In humans, the type I IFNs consist of 13 α-genes coding for 12 IFN-α subtypes, one β-gene encoding a single IFN-β subtype and a single gene encoding IFN-ω. Type I IFNs bind to IFN-αβR, which is a heterodimer of IFN-αR1 and IFN-αR2 chains. High levels of IFN-α subtypes are rapidly secreted from pDCs upon viral recognition, whereas IFN-β can be induced from most virus-infected cell types.
Type I IFN Induction
As described in detail above, stimulation of various PRRs including TLRs (TLR3, 4, 7, 8, and 9), RLRs (RIG-I, MDA-5), and cytosolic DNA sensor(s) leads to the production of type I IFNs. Most virally infected cells trigger the cell intrinsic pathway of type I IFN production through activation of RLRs or DNA sensors. In contrast, pDCs recognize viral genomes within the endosome via TLR7 or TLR9 and induce robust secretion of IFN-α and IFN-β. pDCs are thought to express high constitutive levels of IRF7 and are the only cells capable of coupling TLRs to IRF7 directly, thereby leading to rapid and robust transcription of type I IFN genes.
Amplification of IFN Production by IRF7
In the cytosol of most cells, IRF3 is constitutively expressed while very low levels of IRF7 are found.71 While IFN-α genes contain ISREs, the IFN-β gene also contains binding sites for NF-κB and activator protein 1 (AP1). IRF3 is a potent activator of the IFN-β and IFN-α4 gene but not the IFN-α genes, whereas IRF7 efficiently activates both IFN-α and IFN-β genes. Thus, upon activation of RLRs or TLR3, IFN-β and IFN-α4 production is immediately triggered by the IRF3 homodimer binding to the promoter regions of these genes, while the production of other members of IFN-α occurs at a later time point upon IFN-αβR-induced production of IRF7. In addition, IFN-αβR signaling increases PRR expression (TLRs and RLRs), leading to further amplification of antiviral signaling pathways.
IFN-αβR Signaling
The IFN-αβR utilizes the so-called JAK-STAT signaling pathway, which is used by many other cytokine receptors. This pathway consists of a receptor, JAK family tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2), and transcription factors (STAT1–6). The receptor complex typically consists of two or three distinct polypeptides that contain in their cytoplasmic domain binding sites for different combinations of JAK and STAT proteins. Upon receptor engagement by its ligand, the receptor complex is assembled, resulting in activation of JAK kinases with subsequent tyrosine phosphorylation of STAT proteins, leading to their dimerization, nuclear translocation, and activation of target genes (Fig. 8.6).
Thus, upon engagement of Type I IFN receptor complex IFN-αR1 and IFN-αR2 by IFN-α, IFN-β or IFN-ω, these receptor subunits dimerize, resulting in phosphorylation of Tyk2, which is associated with the IFN-αR1, by Janus kinase (JAK) 2. Activated Tyk2 subsequently phosphorylates JAK1, which is coupled to the IFN-αR2 chain. Signal transducer and activator of transcription (STAT) 1 and 2 are prebound to the IFN-αR2 chain. Activated JAK1 binds to STAT2 and phosphorylates it, creating a binding site for STAT1, causing their dimerization and transport to the nucleus. The STAT1–2 heterodimer forms a transcription complex, ISGF3, with IRF9 to facilitate transcription of ISGs by binding to the sequence motif called interferon simulated response element (ISRE). In addition, STAT1 homodimers bind to the IFN-γ-activated site (GAS) sequence motif and induce a different set of ISGs (Fig. 8.6).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


