55
Immunopharmacology
CASE STUDY
A 30-year-old woman has one living child, age 6. Her child and her husband are Rh positive and she is Rho(D) and Du negative. She is now in her ninth month of pregnancy and is in the labor room having frequent contractions. Her Rh antibody test taken earlier in the pregnancy was negative. What immunotherapy is appropriate for this patient? When and how should it be administered?
Agents that suppress the immune system play an important role in preventing the rejection of organ or tissue grafts and in the treatment of certain diseases that arise from dysregulation of the immune response. While precise details of the mechanisms of action of a number of these agents are still obscure, knowledge of the elements of the immune system is useful in understanding their effects. Agents that augment the immune response or selectively alter the balance of various components of the immune system are also becoming important in the management of certain diseases such as cancer, AIDS, and autoimmune or inflammatory diseases. A growing number of other conditions (infections, cardiovascular diseases, organ transplantation) may also be candidates for immune manipulation.
 ELEMENTS OF THE IMMUNE SYSTEM
ELEMENTS OF THE IMMUNE SYSTEM
NORMAL IMMUNE RESPONSES
The immune system has evolved to protect the host from invading pathogens and to eliminate disease. When functioning at its best, the immune system is exquisitely responsive to invading pathogens while retaining the capacity to recognize self tissues and antigens to which it is tolerant. Protection from infection and disease is provided by the collaborative efforts of the innate and adaptive immune systems.
The Innate Immune System
The innate immune system is the first line of defense against invading pathogens (eg, bacteria, viruses, fungi, parasites) and consists of mechanical, biochemical, and cellular components. Mechanical components include skin/epidermis and mucus; biochemical components include antimicrobial peptides and proteins (eg, defensins), complement, enzymes (eg, lysozyme, acid hydrolases), interferons, acidic pH, and free radicals (eg, hydrogen peroxide, superoxide anions); cellular components include neutrophils, monocytes, macrophages, natural killer (NK), and natural killer-T (NKT) cells. Unlike adaptive immunity, the innate immune response exists prior to infection, is not enhanced by repeated infection, and is generally not antigen-specific. An intact skin or mucosa is the first barrier to infection. When this barrier is breached, an immediate innate immune response, referred to as “inflammation” is provoked that ultimately leads to destruction of the pathogen. The process of pathogen destruction can be accomplished, for example, by biochemical components such as lysozyme (which breaks down bacterial peptidoglycan cell walls) and complement activation. Complement components (Figure 55–1) enhance macrophage and neutrophil phagocytosis by acting as opsonins (C3b) and chemoattractants (C3a, C5a), which recruit immune cells from the bloodstream to the site of infection. The activation of complement eventually leads to pathogen lysis via the generation of a membrane attack complex that creates holes in the pathogen membrane, killing it.
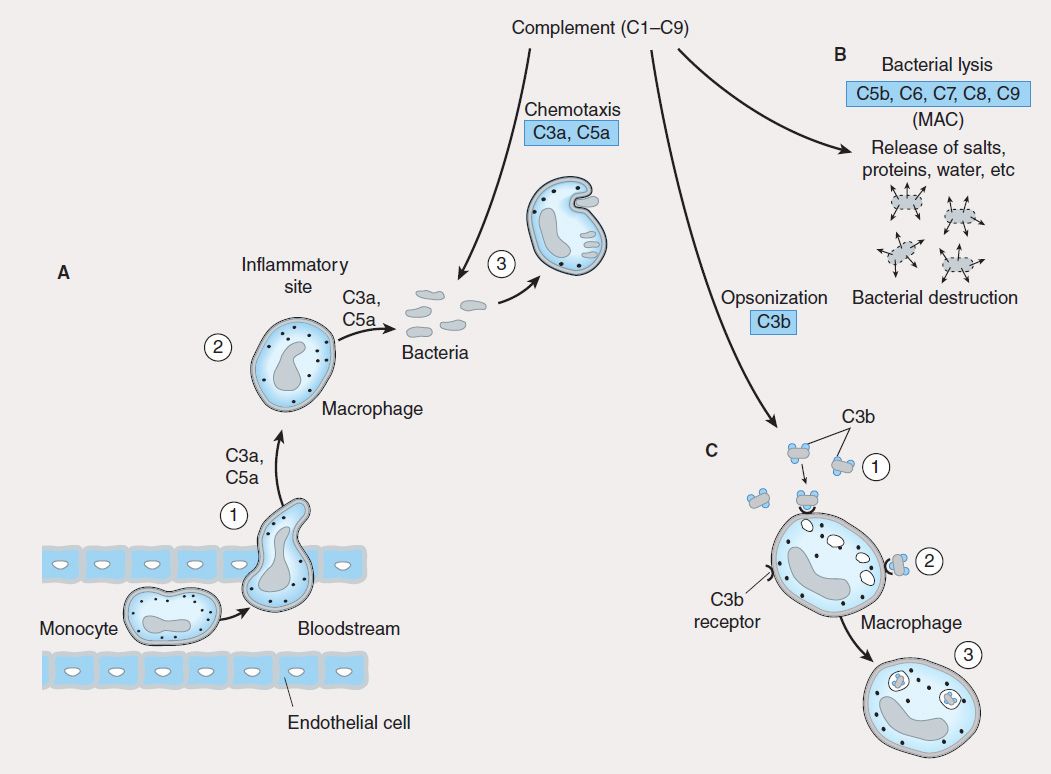
FIGURE 55–1 Role of complement in innate immunity. Complement is made up of nine proteins (C1–C9), which are split into fragments during activation. A: Complement components (C3a, C5a) attract phagocytes (1) to inflammatory sites (2), where they ingest and degrade pathogens (3). B: Complement components C5b, C6, C7, C8, and C9 associate to form a membrane attack complex (MAC) that lyses bacteria, causing their destruction. C: Complement component C3b is an opsonin that coats bacteria (1) and facilitates their ingestion (2) and digestion (3) by phagocytes.
During the inflammatory response triggered by infection, neutrophils and monocytes enter the tissue sites from the peripheral circulation. This cellular influx is mediated by the action of chemoattractant cytokines (chemokines) (eg, interleukin-8 [IL-8; CXCL8], macrophage chemotactic protein-1 [MCP-1; CCL2], and macrophage inflammatory protein-1α [MIP-1α CCL3]) released from activated endothelial cells and immune cells (mostly tissue macrophages) at the inflammatory site. Egress of the immune cells from blood vessels into the inflammatory site is mediated by adhesive interactions between cell surface receptors (eg, L-selectin, integrins) on the immune cells and ligands (eg, sialyl-Lewis x, intercellular adhesion molecule-1 [ICAM-1]) on the activated endothelial cell surface. The tissue macrophages as well as dendritic cells express pattern recognition receptors (PRRs) that include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), scavenger receptors, mannose receptors, and lipopolysaccharide (LPS)-binding protein, which recognize key evolutionarily conserved pathogen components referred to as pathogen-associated molecular patterns (PAMPs). Examples of PAMPs include microbe-derived unmethylated CpG DNA, flagellin, double-stranded RNA, peptidoglycan, and LPS. The PRRs recognize PAMPs in various components of pathogens and stimulate the release of proinflammatory cytokines, chemokines, and interferons. If the innate immune response is successfully executed, the invading pathogen is ingested, degraded, and eliminated, and disease is either prevented or is of short duration.
ACRONYMS
In addition to monocytes and neutrophils, natural killer (NK), natural killer-T (NKT), and gamma-delta T (γδT) cells recruited to the inflammatory site contribute to the innate response by secreting interferon-gamma (IFN-γ) and interleukin-17 (IL-17),* which activate resident tissue macrophages and dendritic cells and recruit neutrophils respectively to successfully eliminate invading pathogens. NK cells are so called because they are able to recognize and destroy virus-infected normal cells as well as tumor cells without prior stimulation. This activity is regulated by “killer cell immunoglobulin-like receptors” (KIRs) on the NK cell surface that are specific for major histocompatibility complex (MHC) class I molecules. When NK cells bind self MHC class I proteins (expressed on all nucleated cells), these receptors deliver inhibitory signals, preventing them from killing normal host cells. Tumor cells or virus-infected cells that have down-regulated MHC class I expression do not engage these KIRs, resulting in activation of NK cells and subsequent destruction of the target cell. NK cells kill target cells by releasing cytotoxic granules such as perforins and granzymes that induce programmed cell death.
NKT cells express T-cell receptors as well as receptors commonly found on NK cells. NKT cells recognize microbial lipid antigens presented by a unique class of MHC-like molecules known as CD1 and have been implicated in host defense against microbial agents, autoimmune diseases, and tumors.
The Adaptive Immune System
The adaptive immune system is mobilized by cues from the innate response when the innate processes are incapable of coping with an infection. The adaptive immune system has a number of characteristics that contribute to its success in eliminating pathogens. These include the ability to (1) respond to a variety of antigens, each in a specific manner; (2) discriminate between foreign (“non-self”) antigens (pathogens) and self antigens of the host; and (3) respond to a previously encountered antigen in a learned way by initiating a vigorous memory response. This adaptive response culminates in the production of antibodies, which are the effectors of humoral immunity; and the activation of T lymphocytes, which are the effectors of cell-mediated immunity.
The induction of specific adaptive immunity requires the participation of professional antigen-presenting cells (APCs), which include dendritic cells (DCs), macrophages, and B lymphocytes. These cells play pivotal roles in the induction of an adaptive immune response because of their capacity to phagocytize particulate antigens (eg, pathogens) or endocytose protein antigens, and enzymatically digest them to generate peptides, which are then loaded onto class I or class II MHC proteins and “presented” to the cell surface T-cell receptor (TCR) (Figure 55–2). CD8 T cells recognize class I-MHC peptide complexes while CD4 T cells recognize class II-MHC peptide complexes. At least two signals are necessary for the activation of T cells. The first signal is delivered following engagement of the TCR with peptide-bound MHC molecules. In the absence of a second signal, the T cells become unresponsive (anergic) or undergo apoptosis. The second signal involves binding of costimulatory molecules (CD40, CD80 [also known as B7-1], and CD86 [also known as B7-2]) on the APC to their respective ligands (CD40L for CD40, CD28 for CD80 or CD86). Activation of T cells is regulated via a negative feedback loop involving another molecule known as T-lymphocyte-associated antigen 4 (CTLA-4). Following engagement of CD28 with CD80 or CD86, CTLA-4 in the cytoplasm is mobilized to the cell surface where, because of its higher affinity of binding to CD80 and CD86, it outcompetes or displaces CD28 resulting in suppression of T-cell activation and proliferation. This property of CTLA-4 has been exploited as a strategy for sustaining a desirable immune response such as that directed against cancer. A recombinant humanized antibody (ipilimumab) that binds CTLA-4 prevents its association with CD80/CD86. In so doing, the activated state of T cells is sustained. Recently completed vaccine trials in metastatic melanoma patients receiving anti-CTLA-4 antibody reported objective and durable clinical responses in a few patients. Unfortunately, these beneficial responses were associated with the development of autoimmune toxicity in some patients, raising concern about this approach.
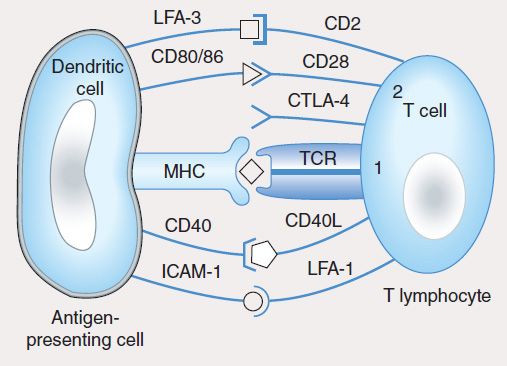
FIGURE 55–2 T-cell activation by an antigen-presenting cell requires engagement of the T-cell receptor by the MHC-peptide complex (signal 1) and binding of the costimulatory molecules (CD80, CD86) on the dendritic cell to CD28 on the T cell (signal 2). The activation signals are strengthened by CD40/CD40L and ICAM-1/LFA-1 interactions. In a normal immune response, T-cell activation is regulated by T-cell-derived CTLA-4, which binds to CD80 or CD86 with higher affinity than CD28 and sends inhibitory signals to the nucleus of the T cell.
T lymphocytes develop and learn to recognize self and non-self antigens in the thymus; those T cells that bind with high affinity to self antigens in the thymus undergo apoptosis (negative selection), while those that are capable of recognizing foreign antigens in the presence of self MHC molecules are retained and expanded (positive selection) for export to the periphery (lymph nodes, spleen, mucosa-associated lymphoid tissue, peripheral blood), where they become activated after encountering MHC-presented peptides (Figures 55–2 and 55–3).
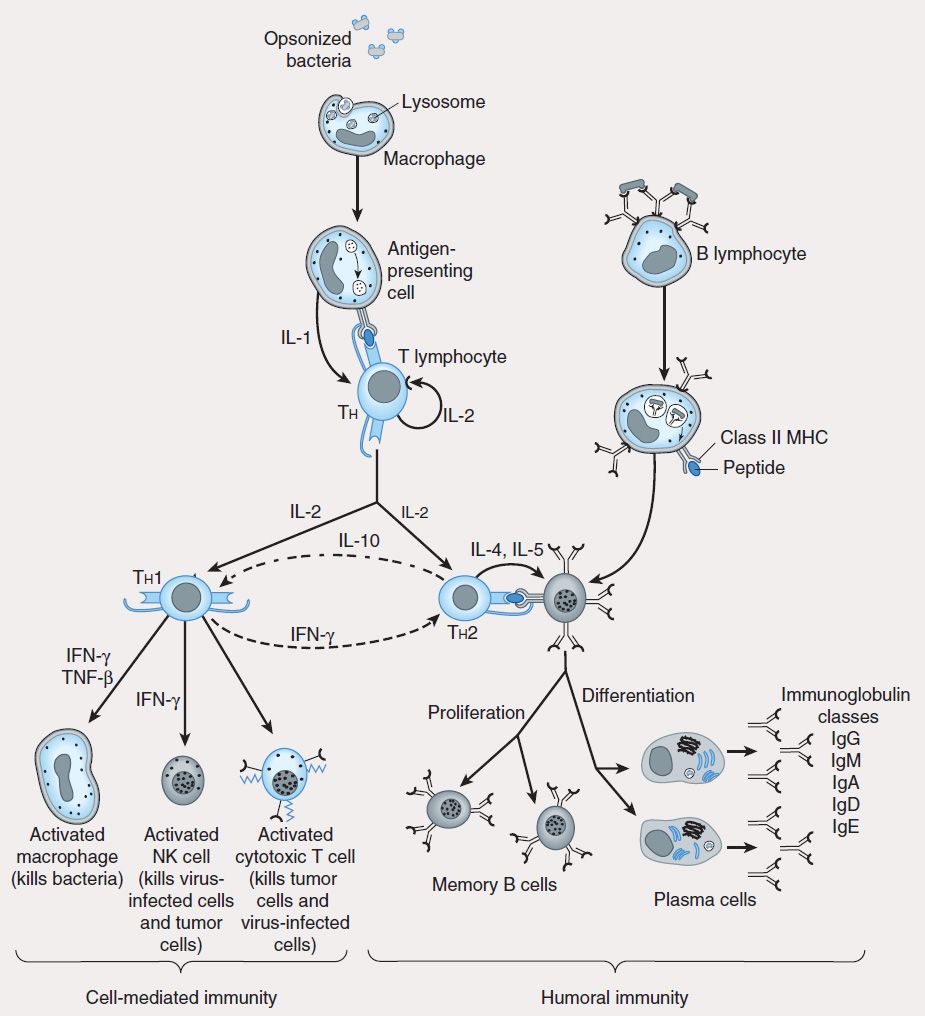
FIGURE 55–3 Scheme of cellular interactions during the generation of cell-mediated and humoral immune responses (see text). The cell-mediated arm of the immune response involves the ingestion and digestion of antigen by antigen-presenting cells such as macrophages. Activated Th cells secrete IL-2, which causes proliferation and activation of cytotoxic T lymphocytes, and Th1 and Th2 cell subsets. Th1 cells also produce IFN-™ and TNF-™, which can directly activate macrophages and NK cells. The humoral response is triggered when B lymphocytes bind antigen via their surface immunoglobulin. They are then induced by Th2-derived IL-4 and IL-5 to proliferate and differentiate into memory cells and antibody-secreting plasma cells. Regulatory cytokines such as IFN-γ and IL-10 down-regulate Th2 and Th1 responses, respectively.
Studies using murine T-cell clones have demonstrated the presence of two subsets of T helper lymphocytes (Th1 and Th2) based on the cytokines they secrete after activation. The TH1 subset characteristically produces IFN-γ, IL-2, and IL-12 and induces cell-mediated immunity by activation of macrophages, cytotoxic T cells (CTLs), and NK cells. The TH2 subset produces IL-4, IL-5, IL-6, and IL-10 (and sometimes IL-13), which induce B-cell proliferation and differentiation into antibody-secreting plasma cells. IL-10 produced by TH2 cells inhibits cytokine production by TH1 cells via the down-regulation of MHC expression by APCs. Conversely, IFN-γ produced by TH1 cells inhibits the proliferation of TH2 cells (Figure 55–3). Although these subsets have been well described in vitro, the nature of the antigenic challenge that elicits a TH1 or TH2 phenotype is less clear. Extracellular bacteria typically cause the elaboration of TH2 cytokines, culminating in the production of neutralizing or opsonic antibodies. In contrast, intracellular organisms (eg, mycobacteria) elicit the production of TH1 cytokines, which lead to the activation of effector cells such as macrophages. A less well-defined T-cell subset (TH3) has been described that produces transforming growth factor-β (TGF-β), whose numerous functions include down-regulation of proliferation and differentiation of T lymphocytes.
Recently, a subset of CD4 T cells that secrete IL-17 (TH17) has been implicated in neutrophil recruitment to sites of inflammation. The regulatory T (Treg) cells comprise a population of CD4 T cells that is essential for preventing autoimmunity and allergy as well as maintaining homeostasis and tolerance to self antigens. This cell population exists as natural Treg (nTreg), derived directly from the thymus, and induced (adaptive) Treg (iTreg), generated from naïve CD4 T cells in the periphery. Both populations have also been shown to inhibit antitumor immune responses and are implicated in fostering tumor growth and progression. Recent attempts to distinguish both populations have resulted in the discovery of a transcription factor, Helios, in nTreg but not in iTreg cells.
CD8 T lymphocytes recognize endogenously processed peptides presented by virus-infected cells or tumor cells. These peptides are usually nine-amino-acid fragments derived from virus or protein tumor antigens in the cytoplasm and are loaded onto MHC class I molecules (Figure 55–2) in the endoplasmic reticulum. In contrast, class II MHC molecules present peptides (usually 11–22 amino acids) derived from extracellular (exogenous) pathogens to CD4 T helper cells. In some instances, exogenous antigens, upon ingestion by APCs, can be presented on class I MHC molecules to CD8 T cells. This phenomenon, referred to as “cross-presentation,” involves retro-translocation of antigens from the endosome to the cytosol for peptide generation in the proteosome and is thought to be useful in generating effective immune responses against infected host cells that are incapable of priming T lymphocytes. Upon activation, CD8 T cells induce target cell death via lytic granule enzymes (“granzymes”), perforin, and the Fas-Fas ligand (Fas-FasL) apoptosis pathways.
B lymphocytes undergo selection in the bone marrow, during which self-reactive B lymphocytes are clonally deleted while B-cell clones specific for foreign antigens are retained and expanded. The repertoire of antigen specificities by T cells is genetically determined and arises from T-cell receptor gene rearrangement while the specificities of B cells arise from immunoglobulin gene rearrangement; for both types of cells, these determinations occur prior to encounters with antigen. Upon an encounter with antigen, a mature B cell binds the antigen, internalizes and processes it, and presents its peptide—bound to class II MHC—to CD4 helper cells, which in turn secrete IL-4 and IL-5. These interleukins stimulate B-cell proliferation and differentiation into memory B cells and antibody-secreting plasma cells. The primary antibody response consists mostly of IgM-class immunoglobulins. Subsequent antigenic stimulation results in a vigorous “booster” response accompanied by class (isotype) switching to produce IgG, IgA, and IgE antibodies with diverse effector functions (Figure 55–3). These antibodies also undergo affinity maturation, which allows them to bind more efficiently to the antigen. With the passage of time, this results in accelerated elimination of microorganisms in subsequent infections. Antibodies mediate their functions by acting as opsonins to enhance phagocytosis and cellular cytotoxicity and by activating complement to elicit an inflammatory response and induce bacterial lysis (Figure 55–4).
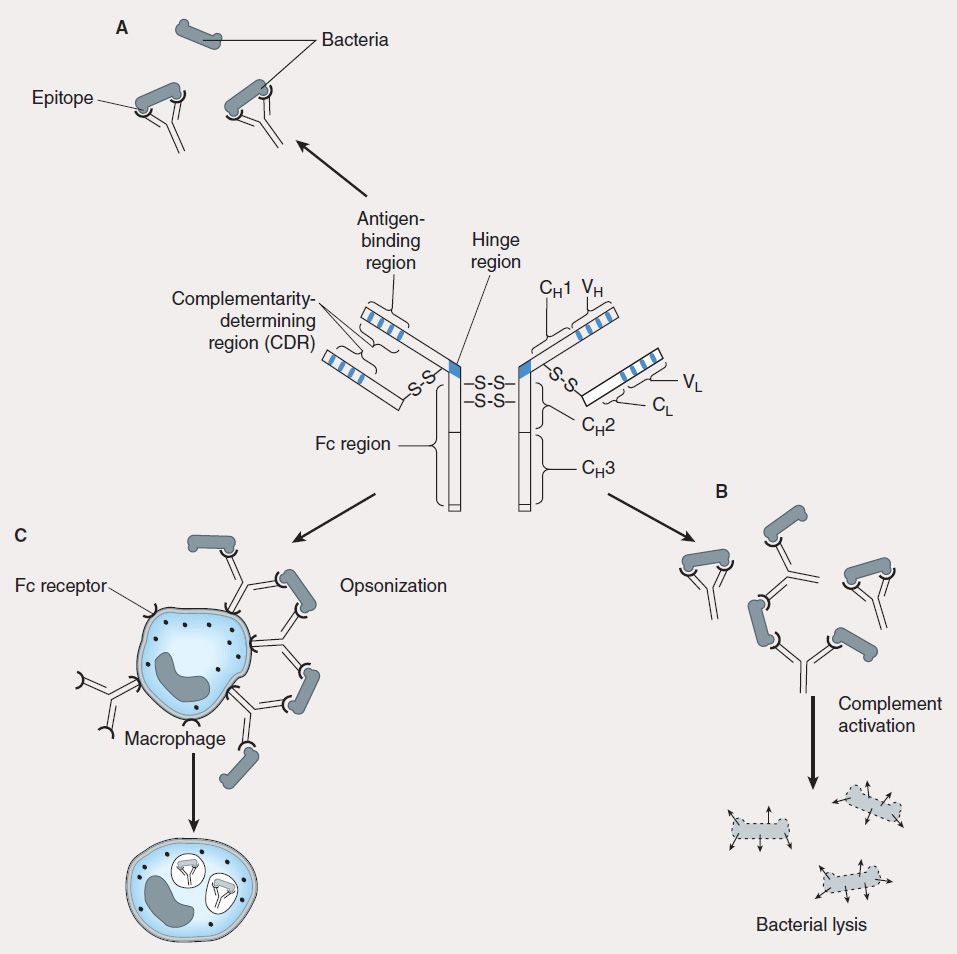
FIGURE 55–4 Antibody has multiple functions. The prototypical antibody consists of two heavy (H) and two light (L) chains, each subdivided into constant (CL, CH) and variable (VL, VH) domains. The structure is held together by intra- and interchain disulfide bridges. A: The complementarity-determining region (CDR) of the antigen-binding portion of the antibody engages the antigenic determinant (epitope) in a lock and key fashion. B: Antigen-antibody complexes activate complement to produce split complement components that cause bacterial lysis. C: The Fc portion of antibodies binds to Fc receptors on phagocytes (eg, macrophages, neutrophils) and facilitates uptake of bacteria (opsonization).
ABNORMAL IMMUNE RESPONSES
Whereas the normally functioning immune response can successfully neutralize toxins, inactivate viruses, destroy transformed cells, and eliminate pathogens, inappropriate responses can lead to extensive tissue damage (hypersensitivity) or reactivity against self antigens (autoimmunity); conversely, impaired reactivity to appropriate targets (immunodeficiency) may occur and abrogate essential defense mechanisms.
Hypersensitivity
Hypersensitivity can be classified as antibody-mediated or cell-mediated. Three types of hypersensitivity are antibody-mediated (types I–III), while the fourth is cell-mediated (type IV). Hypersensitivity occurs in two phases: the sensitization phase and the effector phase. Sensitization occurs upon initial encounter with an antigen; the effector phase involves immunologic memory and results in tissue pathology upon a subsequent encounter with that antigen.
1. Type I—Immediate, or type I, hypersensitivity is IgE-mediated, with symptoms usually occurring within minutes following the patient’s reencounter with antigen. Type I hypersensitivity results from cross-linking of membrane-bound IgE on blood basophils or tissue mast cells by antigen. This cross-linking causes cells to degranulate, releasing substances such as histamine, leukotrienes, and eosinophil chemotactic factor, which induce anaphylaxis, asthma, hay fever, or urticaria (hives) in affected individuals (Figure 55–5). A severe type I hypersensitivity reaction such as systemic anaphylaxis (eg, from insect envenomation, ingestion of certain foods, or drug hypersensitivity) requires immediate medical intervention.
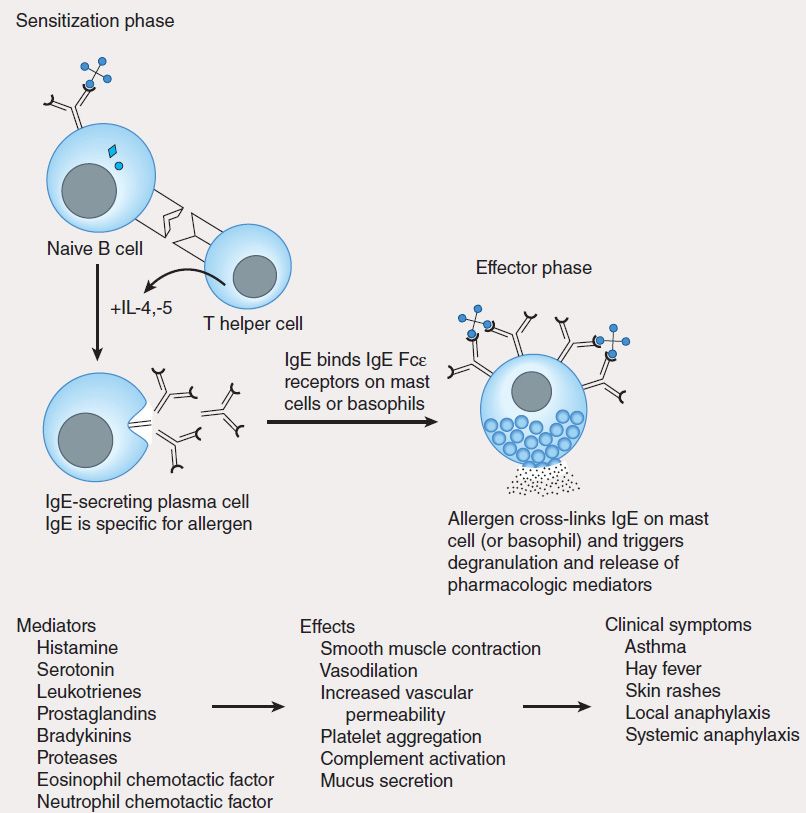
FIGURE 55–5 Mechanism of type I hypersensitivity. Initial exposure to allergen (sensitization phase) leads to production of IgE by plasma cells differentiated from allergen-specific B cells (not shown). The secreted IgE binds IgE-specific receptors (Fc?R) on blood basophils and tissue mast cells. Re-exposure to allergen leads to cross-linking of membrane-bound IgE (effector phase). This cross-linking causes degranulation of cytoplasmic granules and release of mediators that induce vasodilation, smooth muscle contraction, and increased vascular permeability. These effects lead to the clinical symptoms characteristic of type I hypersensitivity.
2. Type II—Type II hypersensitivity results from the formation of antigen-antibody complexes between foreign antigen and IgM or IgG immunoglobulins. One example of this type of hypersensitivity is a blood transfusion reaction that can occur if blood is not cross-matched properly. Preformed antibodies bind to red blood cell membrane antigens that activate the complement cascade, generating a membrane attack complex that lyses the transfused red blood cells. In hemolytic disease of the newborn, anti-Rh IgG antibodies produced by an Rh-negative mother cross the placenta, bind to red blood cells of an Rh-positive fetus, and damage them. The disease is prevented in subsequent pregnancies by the administration of anti-Rh antibodies to the mother 24–48 hours after delivery (see Immunosuppressive Antibodies, below). Type II hypersensitivity can also be drug-induced and may occur during the administration of penicillin (for example) to allergic patients. In these patients, penicillin binds to red blood cells or other host tissue to form a neoantigen that evokes production of antibodies capable of inducing complement-mediated red cell lysis. In some circumstances, subsequent administration of the drug can lead to systemic anaphylaxis (type I hypersensitivity).
3. Type III—Type III hypersensitivity is due to the presence of elevated levels of antigen-antibody complexes in the circulation that ultimately deposit on basement membranes in tissues and vessels. Immune complex deposition activates complement to produce components with anaphylatoxic and chemotactic activities (C5a, C3a, C4a) that increase vascular permeability and recruit neutrophils to the site of complex deposition. Complex deposition and the action of lytic enzymes released by neutrophils can cause skin rashes, glomerulonephritis, and arthritis in these individuals. If patients have type III hypersensitivity against a particular antigen, clinical symptoms usually occur 3–4 days after exposure to the antigen.
4. Type IV: Delayed-type hypersensitivity—Unlike type I, II, and III hypersensitivities, delayed-type hypersensitivity (DTH) is cell-mediated, and responses occur 2–3 days after exposure to the sensitizing antigen. DTH is caused by antigen-specific DTH TH1 cells and induces a local inflammatory response that causes tissue damage characterized by the influx of antigen-nonspecific inflammatory cells, especially macrophages. These cells are recruited under the influence of TH1-produced cytokines (Figure 55–6), which chemoattract circulating monocytes and neutrophils, induce myelopoiesis, and activate macrophages. The activated macrophages are primarily responsible for the tissue damage associated with DTH. Although widely considered to be deleterious, DTH responses are very effective in eliminating infections caused by intracellular pathogens such as Mycobacterium tuberculosis and Leishmania species. Clinical manifestations of DTH include tuberculin and contact hypersensitivities. Tuberculosis exposure is determined using a DTH skin test. Positive responses show erythema and induration caused by accumulation of macrophages and DTH T (TDTH) cells at the site of the tuberculin injection. Poison ivy is the most common cause of contact hypersensitivity, in which pentadecacatechol, the lipophilic chemical in poison ivy, modifies cellular tissue and results in a DTH T-cell response.
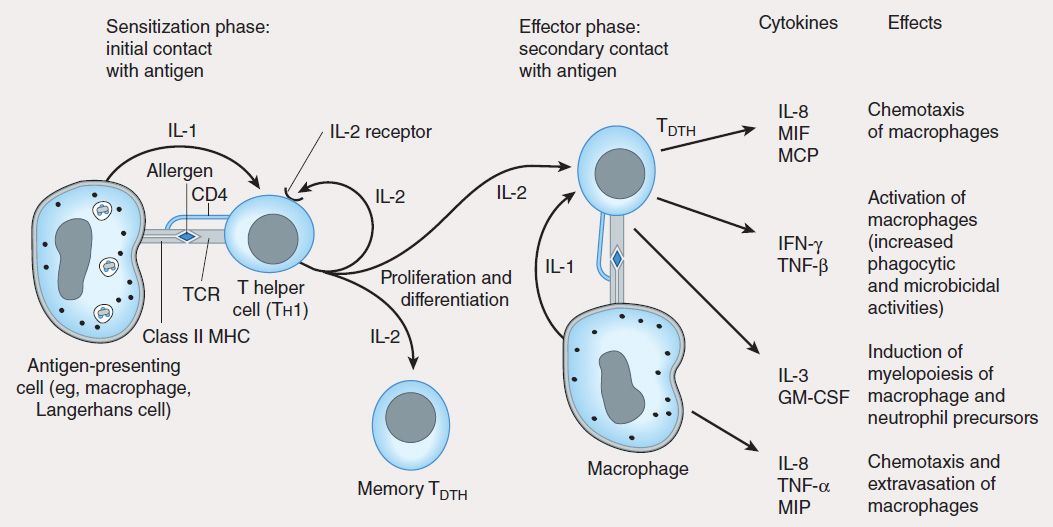
FIGURE 55–6 Mechanism of type IV hypersensitivity (DTH). In the sensitization phase, the processed allergen (eg, from poison ivy) is presented to CD4 Th1 cells by antigen-presenting cells in association with class II MHC. T cells are induced to express IL-2 receptors and are stimulated to proliferate and differentiate into memory TDTH cells. Secondary contact with antigen triggers the effector phase, in which memory TDTH cells release cytokines that attract and activate nonspecific inflammatory macrophages and neutrophils. These cells display increased phagocytic and microbicidal activities and release large quantities of lytic enzymes that cause extensive tissue damage.
Autoimmunity
Autoimmune disease arises when the body mounts an immune response against itself due to failure to distinguish self tissues and cells from foreign (nonself) antigens or loss of tolerance to self. This phenomenon derives from the activation of self-reactive T and B lymphocytes that generate cell-mediated or humoral immune responses directed against self antigens. The pathologic consequences of this reactivity constitute several types of autoimmune diseases. Autoimmune diseases are highly complex due to MHC genetics, environmental conditions, infectious entities, and dysfunctional immune regulation. Examples of such diseases include rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, and insulin-dependent diabetes mellitus (type 1 diabetes). In rheumatoid arthritis, IgM antibodies (rheumatoid factors) are produced that react with the Fc portion of IgG and may form immune complexes that activate the complement cascade, causing chronic inflammation of the joints and kidneys. In systemic lupus erythematosus, antibodies are made against DNA, histones, red blood cells, platelets, and other cellular components. In multiple sclerosis and type 1 diabetes, cell-mediated autoimmune attack destroys myelin surrounding nerve cells and insulin-producing islet beta cells of the pancreas, respectively. In type 1 diabetes, activated CD4 TDTH cells that infiltrate the islets of Langerhans and recognize self islet beta cell peptides are thought to produce cytokines that stimulate macrophages to produce lytic enzymes, which destroy islet beta cells. Autoantibodies directed against the islet beta cell antigens are produced, but do not contribute significantly to disease.
A number of mechanisms have been proposed to explain autoimmunity:
1. Exposure of antigens previously sequestered from the immune system (eg, lens protein, myelin basic protein) to self-reactive T lymphocytes.
2. Molecular mimicry by invading pathogens, in which immune responses are directed at antigenic determinants on pathogens that share identical or similar epitopes with normal host tissue. This phenomenon occurs in rheumatic fever following Streptococcus pyogenes infection, in which heart damage is thought to arise from an immune response directed against streptococcal antigens shared with heart muscle. The suggested viral etiology of autoimmune diseases has been ascribed to immune responses (both cell-mediated and humoral) directed against virus epitopes that mimic self antigens.
3. Inappropriate expression of class II MHC molecules on the membranes of cells that normally do not express class II MHC (eg, islet beta cells). Increased expression of MHC II may increase presentation of self peptides to T helper cells, which in turn induce CTL, TDTH, and B-lymphocyte cells that react against self antigens.
Immunodeficiency Diseases
Immunodeficiency diseases result from inadequate function in the immune system; the consequences include increased susceptibility to infections and prolonged duration and severity of disease. Immunodeficiency diseases are either congenital or arise from extrinsic factors such as bacterial or viral infections or drug treatment. Affected individuals frequently succumb to infections caused by opportunistic organisms of low pathogenicity for the immunocompetent host. Examples of congenitally acquired immunodeficiency diseases include X-linked agammaglobulinemia, DiGeorge’s syndrome, and severe combined immunodeficiency disease (SCID) due to adenosine deaminase (ADA) deficiency.
X-linked agammaglobulinemia is a disease affecting males that is characterized by a failure of immature B lymphocytes to mature into antibody-producing plasma cells. These individuals are susceptible to recurrent bacterial infections, although the cell-mediated responses directed against viruses and fungi are preserved. DiGeorge’s syndrome is due to failure of the thymus to develop, resulting in diminished T-cell responses (TDTH, CTL), while the humoral response remains functional, but does not benefit from T-cell help.
The ADA enzyme normally prevents the accumulation of toxic deoxy-ATP in cells. Deoxy-ATP is particularly toxic to lymphocytes, and leads to death of T and B cells. Absence of the enzyme therefore results in SCID. Infusion of the purified enzyme (pegademase, from bovine sources) and transfer of ADA gene-modified lymphocytes have both been used successfully to treat this disease.
AIDS represents the classic example of immunodeficiency disease caused by extrinsic viral infection, in this instance the human immunodeficiency virus (HIV). This virus exhibits a strong tropism for CD4 T helper cells; these become depleted, giving rise to increased frequency of opportunistic infections and malignancies in infected individuals. AIDS is also characterized by an imbalance in TH1 and TH2 cells, and the ratios of cells and their functions are skewed toward TH2. This results in loss of cytotoxic T-lymphocyte activity, loss of delayed hypersensitivity, and hypergammaglobulinemia.
 IMMUNOSUPPRESSIVE THERAPY
IMMUNOSUPPRESSIVE THERAPY
Immunosuppressive agents have proved very useful in minimizing the occurrence or impact of deleterious effects of exaggerated or inappropriate immune responses. Unfortunately, these agents also have the potential to cause disease and to increase the risk of infection and malignancies.
GLUCOCORTICOIDS
Glucocorticoids (corticosteroids) were the first hormonal agents recognized as having lympholytic properties. Administration of any glucocorticoid reduces the size and lymphoid content of the lymph nodes and spleen, although it has no toxic effect on proliferating myeloid or erythroid stem cells in the bone marrow.
Glucocorticoids are thought to interfere with the cell cycle of activated lymphoid cells. The mechanism of their action is described in Chapter 39. Glucocorticoids are quite cytotoxic to certain subsets of T cells, but their immunologic effects are probably due to their ability to modify cellular functions rather than to direct cytotoxicity. Although cellular immunity is more affected than humoral immunity, the primary antibody response can be diminished, and with continued use, previously established antibody responses are also decreased. Additionally, continuous administration of corticosteroid increases the fractional catabolic rate of IgG, the major class of antibody immunoglobulins, thus lowering the effective concentration of specific antibodies. Contact hypersensitivity mediated by DTH T cells, for example, is usually abrogated by glucocorticoid therapy.
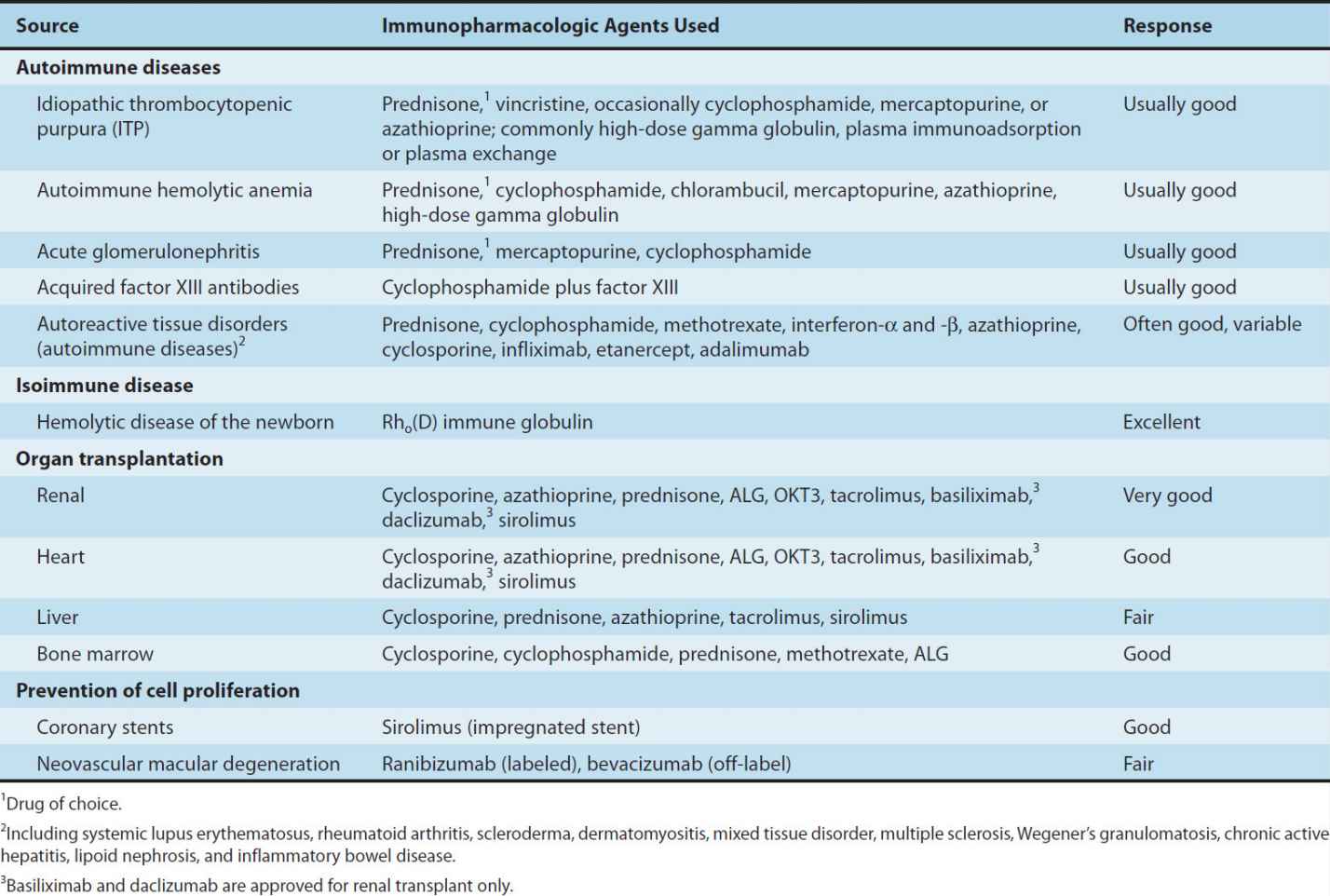
Glucocorticoids are used in a wide variety of conditions (Table 55–1). It is thought that the immunosuppressive and anti-inflammatory properties of corticosteroids account for their beneficial effects in diseases like idiopathic thrombocytopenic purpura and rheumatoid arthritis. Glucocorticoids modulate allergic reactions and are useful in the treatment of diseases like asthma or as premedication for other agents (eg, blood products, chemotherapy) that might cause undesirable immune responses. Glucocorticoids are first-line immunosuppressive therapy for both solid organ and hematopoietic stem cell transplant recipients, with variable results. The toxicities of long-term glucocorticoid therapy can be severe and are discussed in Chapter 39.
TABLE 55–1 Clinical uses of immunosuppressive agents.
CALCINEURIN INHIBITORS
Cyclosporine
Cyclosporine (cyclosporin A, CSA) is an immunosuppressive agent with efficacy in human organ transplantation, in the treatment of graft-versus-host (GVH) disease after hematopoietic stem cell transplantation, and in the treatment of selected autoimmune disorders. Cyclosporine is a peptide antibiotic that appears to act at an early stage in the antigen receptor-induced differentiation of T cells and blocks their activation. Cyclosporine binds to cyclophilin, a member of a class of intracellular proteins called immunophilins. Cyclosporine and cyclophilin form a complex that inhibits the cytoplasmic phosphatase, calcineurin, which is necessary for the activation of a T-cell-specific transcription factor. This transcription factor, NF-AT, is involved in the synthesis of interleukins (eg, IL-2) by activated T cells. In vitro studies have indicated that cyclosporine inhibits the gene transcription of IL-2, IL-3, IFN-γ, and other factors produced by antigen-stimulated T cells, but it does not block the effect of such factors on primed T cells nor does it block interaction with antigen.
Cyclosporine may be given intravenously or orally, though it is slowly and incompletely absorbed (20–50%). The absorbed drug is primarily metabolized by the P450 3A enzyme system in the liver with resultant multiple drug interactions. This propensity for drug interactions contributes to significant interpatient variability in bioavailability, such that cyclosporine requires individual patient dosage adjustments based on steady-state blood levels and the desired therapeutic ranges for the drug. Cyclosporine ophthalmic solution is now available for severe dry eye syndrome, as well as ocular GVH disease. Inhaled cyclosporine is being investigated for use in lung transplantation.
Toxicities are numerous and include nephrotoxicity, hypertension, hyperglycemia, liver dysfunction, hyperkalemia, altered mental status, seizures, and hirsutism. Cyclosporine causes very little bone marrow toxicity. While an increased incidence of lymphoma and other cancers (Kaposi’s sarcoma, skin cancer) have been observed in transplant recipients receiving cyclosporine, other immunosuppressive agents may also predispose recipients to cancer. Some evidence suggests that tumors may arise after cyclosporine treatment because the drug induces TGF-β, which promotes tumor invasion and metastasis.
Cyclosporine may be used alone or in combination with other immunosuppressants, particularly glucocorticoids. It has been used successfully as the sole immunosuppressant for cadaveric transplantation of the kidney, pancreas, and liver, and it has proved extremely useful in cardiac transplantation as well. In combination with methotrexate, cyclosporine is a standard prophylactic regimen to prevent GVH disease after allogeneic stem cell transplantation. Cyclosporine has also proved useful in a variety of autoimmune disorders, including uveitis, rheumatoid arthritis, psoriasis, and asthma. Its combination with newer agents is showing considerable efficacy in clinical and experimental settings where effective and less toxic immunosuppression is needed. Newer formulations of cyclosporine have been developed that are improving patient compliance (smaller, better tasting pills), and increasing bioavailability.
Tacrolimus
Tacrolimus (FK 506) is an immunosuppressant macrolide antibiotic produced by Streptomyces tsukubaensis. It is not chemically related to cyclosporine, but their mechanisms of action are similar. Both drugs bind to cytoplasmic peptidylprolyl isomerases that are abundant in all tissues. While cyclosporine binds to cyclophilin, tacrolimus binds to the immunophilin FK-binding protein (FKBP). Both complexes inhibit calcineurin, which is necessary for the activation of the T-cell-specific transcription factor NF-AT.
On a weight basis, tacrolimus is 10–100 times more potent than cyclosporine in inhibiting immune responses. Tacrolimus is utilized for the same indications as cyclosporine, particularly in organ and stem cell transplantation. Multicenter studies in the USA and in Europe indicate that both graft and patient survival are similar for the two drugs. Tacrolimus has proven to be effective therapy for preventing rejection in solid-organ transplant patients even after failure of standard rejection therapy, including anti-T-cell antibodies. It is now considered a standard prophylactic agent (usually in combination with methotrexate or mycophenolate mofetil) for GVH disease.
Tacrolimus can be administered orally or intravenously. The half-life of the intravenous form is approximately 9–12 hours. Like cyclosporine, tacrolimus is metabolized primarily by P450 enzymes in the liver, and there is potential for drug interactions. The dosage is determined by trough blood level at steady state. Its toxic effects are similar to those of cyclosporine and include nephrotoxicity, neurotoxicity, hyperglycemia, hypertension, hyperkalemia, and gastrointestinal complaints.
Because of the effectiveness of systemic tacrolimus in some dermatologic diseases, a topical preparation is now available. Tacrolimus ointment is currently used in the therapy of atopic dermatitis and psoriasis.
PROLIFERATION SIGNAL INHIBITORS
A new class of immunosuppressive agents called proliferation-signal inhibitors (PSIs) includes sirolimus (rapamycin) and its derivative everolimus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


