37
Hypothalamic & Pituitary Hormones
CASE STUDY
A 3-year-old boy (height 85 cm, –3 standard deviations [SD]; weight 13 kg, approximately 10th percentile) presents with short stature. Review of the past history and growth chart demonstrates normal birth weight and birth length, but a progressive fall off in height velocity relative to age-matched normal ranges starting at 6 months of age. Physical examination demonstrates short stature and mild generalized obesity. Genital examination reveals descended but small testes and a phallic length of –2 SD. Laboratory evaluations demonstrate growth hormone (GH) deficiency and a delayed bone age of 18 months. The patient is started on replacement with recombinant human GH at a dose of 40 mcg/kg/d subcutaneously. After 1 year of treatment, his height velocity has increased from 5 cm/y to 11 cm/y. How does GH stimulate growth in children? What other hormone deficiencies are suggested by the patient’s physical examination? What other hormone supplementation is this patient likely to require?
The control of metabolism, growth, and reproduction is mediated by a combination of neural and endocrine systems located in the hypothalamus and pituitary gland. The pituitary weighs about 0.6 g and rests at the base of the brain in the bony sella turcica near the optic chiasm and the cavernous sinuses. The pituitary consists of an anterior lobe (adenohypophysis) and a posterior lobe (neurohypophysis) (Figure 37–1). It is connected to the overlying hypothalamus by a stalk of neurosecretory fibers and blood vessels, including a portal venous system that drains the hypothalamus and perfuses the anterior pituitary. The portal venous system carries small regulatory hormones (Figure 37–1, Table 37–1) from the hypothalamus to the anterior pituitary.
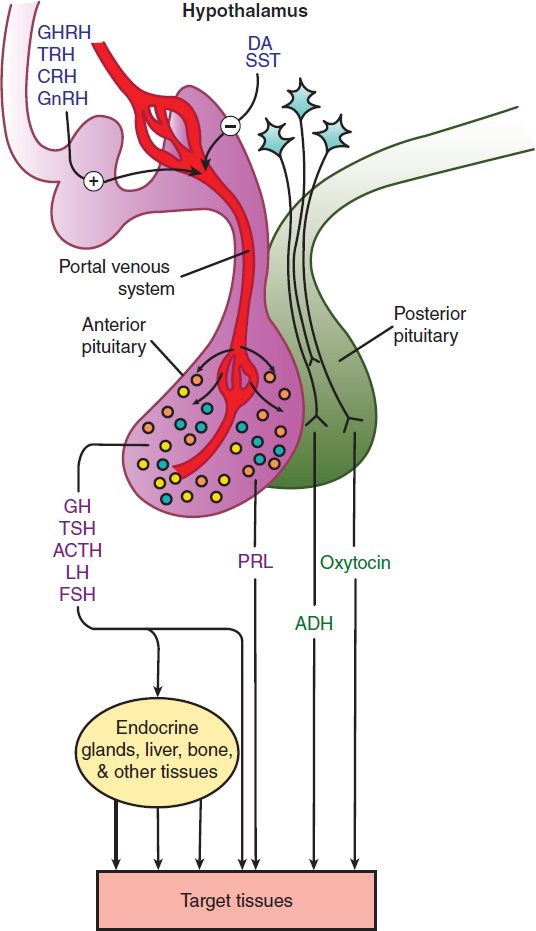
FIGURE 37–1 The hypothalamic-pituitary endocrine system. Hormones released from the anterior pituitary stimulate the production of hormones by a peripheral endocrine gland, the liver, or other tissues, or act directly on target tissues. Prolactin and the hormones released from the posterior pituitary (vasopressin and oxytocin) act directly on target tissues. Hypothalamic factors regulate the release of anterior pituitary hormones. ACTH, adrenocorticotropin; ADH, antidiuretic hormone [vasopressin]; CRH, corticotropin-releasing hormone; DA, dopamine; FSH, follicle-stimulating hormone; GH, growth hormone; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; PRL, prolactin; SST, somatostatin; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
TABLE 37–1 Links between hypothalamic, anterior pituitary, and target organ hormone or mediator.1
The posterior lobe hormones are synthesized in the hypothalamus and transported via the neurosecretory fibers in the stalk of the pituitary to the posterior lobe; from there they are released into the circulation.
Drugs that mimic or block the effects of hypothalamic and pituitary hormones have pharmacologic applications in three primary areas: (1) as replacement therapy for hormone deficiency states; (2) as antagonists for diseases caused by excess production of pituitary hormones; and (3) as diagnostic tools for identifying several endocrine abnormalities.
 ANTERIOR PITUITARY HORMONES & THEIR HYPOTHALAMIC REGULATORS
ANTERIOR PITUITARY HORMONES & THEIR HYPOTHALAMIC REGULATORS
All the hormones produced by the anterior pituitary except prolactin are key participants in hormonal systems in which they regulate the production of hormones and autocrine-paracrine factors by endocrine glands and other peripheral tissues. In these systems, the secretion of the pituitary hormone is under the control of one or more hypothalamic hormones. Each hypothalamic-pituitary-endocrine gland system or axis provides multiple opportunities for complex neuroendocrine regulation of growth and development, metabolism, and reproductive function.
ANTERIOR PITUITARY & HYPOTHALAMIC HORMONE RECEPTORS
The anterior pituitary hormones can be classified according to hormone structure and the types of receptors that they activate. Growth hormone (GH) and prolactin (PRL), single-chain protein hormones with significant homology, form one group. Both hormones activate receptors of the JAK/STAT superfamily (see Chapter 2). Three pituitary hormones—thyroid-stimulating hormone (TSH, thyrotropin), follicle-stimulating hormone (FSH), and luteinizing hormone (LH)—are dimeric proteins that activate G protein-coupled receptors (see Chapter 2). TSH, FSH, and LH share a common α subunit. Their β subunits, though somewhat similar to each other, differ enough to confer receptor specificity. Finally, adrenocorticotropic hormone (ACTH), a single peptide cleaved from a larger precursor, pro-opiomelanocortin (POMC), that can be cleaved into various other biologically active peptides like α-melanocyte-stimulating hormone (MSH) and β-endorphin (see Chapter 31), represents a third category. Like TSH, LH, and FSH, ACTH acts through a G protein-coupled receptor. A unique feature of the ACTH receptor (also known as the melanocortin 2 receptor) is that a transmembrane protein, melanocortin 2 receptor accessory protein, is essential for normal ACTH receptor trafficking and signaling.
ACRONYMS

TSH, FSH, LH, and ACTH share similarities in the regulation of their release from the pituitary. Each is under the control of a distinctive hypothalamic peptide that stimulates their production by acting on G protein-coupled receptors (Table 37–1). TSH release is regulated by thyrotropin-releasing hormone (TRH), whereas the release of LH and FSH (known collectively as gonadotropins) is stimulated by pulses of gonadotropin-releasing hormone (GnRH). ACTH release is stimulated by corticotropin-releasing hormone (CRH). An important regulatory feature shared by these four structurally related hormones is that they and their hypothalamic releasing factors are subject to feedback inhibitory regulation by the hormones whose production they control. TSH and TRH production are inhibited by the two key thyroid hormones, thyroxine and triiodothyronine (see Chapter 38). Gonadotropin and GnRH production is inhibited in women by estrogen and progesterone, and in men by testosterone and other androgens. ACTH and CRH production are inhibited by cortisol. Feedback regulation is critical to the physiologic control of thyroid, adrenal cortical, and gonadal function and is also important in pharmacologic treatments that affect these systems.
The hypothalamic hormonal control of GH and prolactin differs from the regulatory systems for TSH, FSH, LH, and ACTH. The hypothalamus secretes two hormones that regulate GH; growth hormone-releasing hormone (GHRH) stimulates GH production, whereas the peptide somatostatin (SST) inhibits GH production. GH and its primary peripheral mediator, insulin-like growth factor-I (IGF-I), also provide feedback to inhibit GH release. Prolactin production is inhibited by the catecholamine dopamine acting through the D2 subtype of dopamine receptors. The hypothalamus does not produce a hormone that specifically stimulates prolactin secretion, although TRH can stimulate prolactin release, particularly when TRH concentrations are high in the setting of primary hypothyroidism.
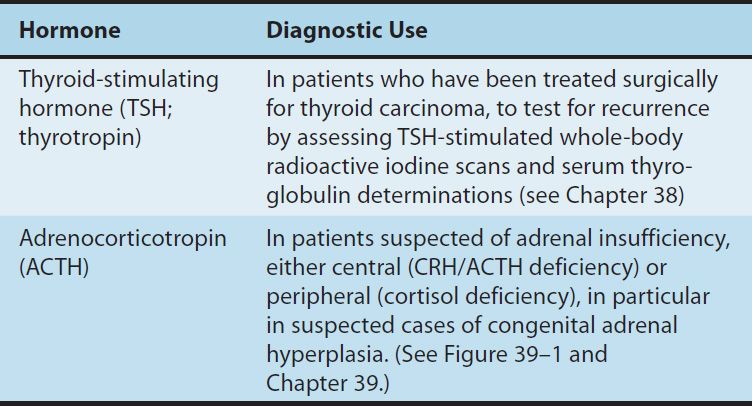
Whereas all the pituitary and hypothalamic hormones described previously are available for use in humans, only a few are of major clinical importance. Because of the greater ease of administration of target endocrine gland hormones or their synthetic analogs, the related hypothalamic and pituitary hormones (TRH, TSH, CRH, ACTH, GHRH) are used infrequently as treatments. Some, such as ACTH, are used for specialized diagnostic testing. These agents are described in Tables 37–2 and 37–3 and are not discussed further in this chapter. In contrast, GH, SST, LH, FSH, GnRH, and dopamine or analogs of these hormones are commonly used and are described in the following text.
TABLE 37–2 Clinical uses of hypothalamic hormones and their analogs.
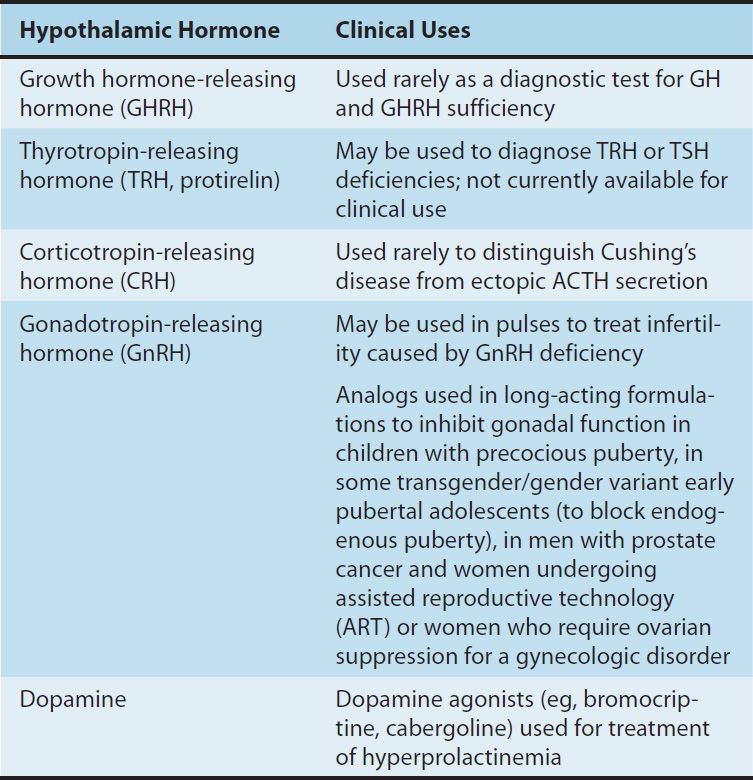
TABLE 37–3 Diagnostic uses of thyroid-stimulating hormone and adrenocorticotropin.
GROWTH HORMONE (SOMATOTROPIN)
Growth hormone, an anterior pituitary hormone, is required during childhood and adolescence for attainment of normal adult size and has important effects throughout postnatal life on lipid and carbohydrate metabolism, and on lean body mass and bone density. Its growth-promoting effects are primarily mediated via IGF-I (also known as somatomedin C). Individuals with congenital or acquired deficiency of GH during childhood or adolescence fail to reach their midparental target adult height and have disproportionately increased body fat and decreased muscle mass. Adults with GH deficiency also have disproportionately low lean body mass.
Chemistry & Pharmacokinetics
A. Structure
Growth hormone is a 191-amino-acid peptide with two sulfhydryl bridges. Its structure closely resembles that of prolactin. In the past, medicinal GH was isolated from the pituitaries of human cadavers. However, this form of GH was found to be contaminated with prions that could cause Creutzfeldt-Jakob disease. For this reason, it is no longer used. Somatropin, the recombinant form of GH, has a 191-amino-acid sequence that is identical with the predominant native form of human GH.
B. Absorption, Metabolism, and Excretion
Circulating endogenous GH has a half-life of approximately 20 minutes and is predominantly cleared by the liver. Recombinant human GH (rhGH) is administered subcutaneously 6–7 times per week. Peak levels occur in 2–4 hours and active blood levels persist for approximately 36 hours.
Pharmacodynamics
Growth hormone mediates its effects via cell surface receptors of the JAK/STAT cytokine receptor superfamily. The hormone has two distinct GH receptor binding sites. Dimerization of two GH receptors is stimulated by a single GH molecule and activates signaling cascades mediated by receptor-associated JAK tyrosine kinases and STATs (see Chapter 2). The hormone has complex effects on growth, body composition, and carbohydrate, protein, and lipid metabolism. The growth-promoting effects are mediated principally, but not solely, through an increase in the production of IGF-I. Much of the circulating IGF-I is produced in the liver. Growth hormone also stimulates production of IGF-I in bone, cartilage, muscle, kidney, and other tissues, where it has autocrine or paracrine roles. It stimulates longitudinal bone growth until the epiphyses close—near the end of puberty. In both children and adults, GH has anabolic effects in muscle and catabolic effects in adipose cells that shift the balance of body mass to an increase in muscle mass and a reduction in adiposity. The direct and indirect effects of GH on carbohydrate metabolism are mixed, in part because GH and IGF-I have opposite effects on insulin sensitivity. Growth hormone reduces insulin sensitivity, which results in mild hyperinsulinemia and increased blood glucose levels, whereas IGF-I has insulin-like effects on glucose transport. In patients who are unable to respond to growth hormone because of severe resistance (caused by GH receptor mutations, post- receptor signaling mutations, or GH antibodies), the administration of recombinant human IGF-I may cause hypoglycemia because of its insulin-like effects.
Clinical Pharmacology
A. Growth Hormone Deficiency
Growth hormone deficiency can have a genetic basis, be associated with midline developmental defect syndromes (eg, septo-optic dysplasia), or be acquired as a result of damage to the pituitary or hypothalamus by a traumatic event (including breech or traumatic delivery), intracranial tumors, infection, infiltrative or hemorrhagic processes, or irradiation. Neonates with isolated GH deficiency are typically of normal size at birth because prenatal growth is not GH-dependent. In contrast, IGF-I is essential for normal prenatal and postnatal growth. Through poorly understood mechanisms, IGF-I expression and postnatal growth become GH-dependent during the first year of life. In childhood, GH deficiency typically presents as short stature, often with mild adiposity. Another early sign of GH deficiency is hypoglycemia due to the loss of a counter-regulatory hormonal response to hypoglycemia; young children are at risk for this condition due to high sensitivity to insulin. Criteria for diagnosis of GH deficiency usually include (1) a subnormal height velocity for age and (2) a subnormal serum GH response following provocative testing with at least two GH secretagogues. Clonidine (α2-adrenergic agonist), levodopa (dopaminergic agonist), and exercise are factors that increase GHRH levels. Arginine and insulin-induced hypoglycemia cause diminished SST, which increases GH release. The prevalence of GH deficiency is approximately 1:5000. If therapy with rhGH is initiated at an early age, many children with short stature due to GH deficiency will achieve an adult height within their midparental target height range.
In the past, it was believed that adults with GH deficiency do not exhibit a significant syndrome. However, more detailed studies suggest that adults with GH deficiency often have generalized obesity, reduced muscle mass, asthenia, diminished bone mineral density, dyslipidemia, and reduced cardiac output. Growth hormone-deficient adults who have been treated with GH experience reversal of many of these manifestations.
B. Growth Hormone Treatment of Pediatric Patients with Short Stature
Although the greatest improvement in growth occurs in patients with GH deficiency, exogenous GH has some effect on height in children with short stature caused by conditions other than GH deficiency. Growth hormone has been approved for several conditions (Table 37–4) and has been used experimentally or off-label in many others. Prader-Willi syndrome is an autosomal dominant genetic disease associated with growth failure, obesity, and carbohydrate intolerance. In children with Prader-Willi syndrome and growth failure, GH treatment decreases body fat and increases lean body mass, linear growth, and energy expenditure.
TABLE 37–4 Clinical uses of recombinant human growth hormone.
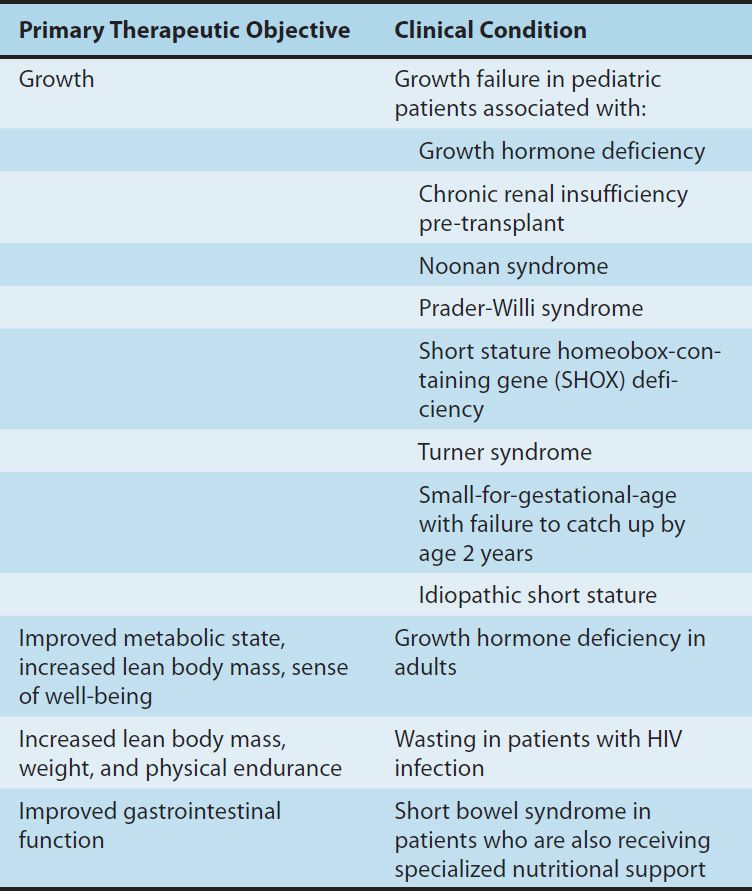
Growth hormone treatment has also been shown to have a strong beneficial effect on final height of girls with Turner syndrome (45 X karyotype and variants). In clinical trials, GH treatment has been shown to increase final height in girls with Turner syndrome by 10–15 cm (4–6 inches). Because girls with Turner syndrome also have either absent or rudimentary ovaries, GH must be judiciously combined with gonadal steroids to achieve maximal height. Other conditions of pediatric growth failure for which GH treatment is approved include chronic renal insufficiency pre-transplant and small-for-gestational-age at birth in which the child’s height remains more than 2 standard deviations below normal at 2 years of age.
A controversial but approved use of GH is for children with idiopathic short stature (ISS). This is a heterogeneous population that has in common no identifiable cause of the short stature. Some have arbitrarily defined ISS clinically as having a height at least 2.25 standard deviations below normal for children of the same age and a predicted adult height that is less than 2.25 standard deviations below normal. In this group of children, many years of GH therapy result in an average increase in adult height of 4–7 cm (1.57–2.76 inches) at a cost of $5000–$40,000 per year. The complex issues involved in the cost-risk-benefit relationship of this use of GH are important because an estimated 400,000 children in the United States fit the diagnostic criteria for ISS.
Treatment of children with short stature should be carried out by specialists experienced in GH administration. Dose requirements vary with the condition being treated, with GH-deficient children typically being most responsive. Children must be observed closely for slowing of growth velocity, which could indicate a need to increase the dosage or the possibility of epiphyseal fusion or intercurrent problems such as hypothyroidism or malnutrition.
Other Uses of Growth Hormone
Growth hormone affects many organ systems and also has a net anabolic effect. It has been tested in a number of conditions that are associated with a severe catabolic state and is approved for the treatment of wasting in patients with AIDS. In 2004, GH was approved for treatment of patients with short bowel syndrome who are dependent on total parenteral nutrition (TPN). After intestinal resection or bypass, the remaining functional intestine in many patients undergoes extensive adaptation that allows it to adequately absorb nutrients. However, other patients fail to adequately adapt and develop a malabsorption syndrome. Growth hormone has been shown to increase intestinal growth and improve its function in experimental animals. Benefits of GH treatment for patients with short bowel syndrome and dependence on TPN have mostly been short-lived in the clinical studies that have been published to date. Growth hormone is administered with glutamine, which also has trophic effects on the intestinal mucosa.
Growth hormone is a popular component of “anti-aging” programs. Serum levels of GH normally decline with aging; anti-aging programs claim that injection of GH or administration of drugs purported to increase GH release are effective anti-aging remedies. These claims are largely unsubstantiated. In contrast, studies in mice and the nematode Caenorhabditis elegans have clearly demonstrated that analogs of human GH and IGF-I consistently shorten life span and that loss-of-function mutations in the signaling pathways for the GH and IGF-I analogs lengthen life span. Another use of GH is by athletes for a purported increase in muscle mass and athletic performance. Growth hormone is one of the drugs banned by the International Olympic Committee.
In 1993, the FDA approved the use of recombinant bovine growth hormone (rbGH) in dairy cattle to increase milk production. Although milk and meat from rbGH-treated cows appear to be safe, these cows have a higher incidence of mastitis, which could increase antibiotic use and result in greater antibiotic residues in milk and meat.
Toxicity & Contraindications
Children generally tolerate growth hormone treatment well. Adverse events are relatively rare and include pseudotumor cerebri, slipped capital femoral epiphysis, progression of scoliosis, edema, hyperglycemia, and increased risk of asphyxiation in severely obese patients with Prader-Willi syndrome and upper airway obstruction or sleep apnea. Patients with Turner syndrome have an increased risk of otitis media while taking GH. In children with GH deficiency, periodic evaluation of the other anterior pituitary hormones may reveal concurrent deficiencies, which also require treatment (ie, with hydrocortisone, levothyroxine, or gonadal hormones). Pancreatitis, gynecomastia, and nevus growth have occurred in patients receiving GH. Adults tend to have more adverse effects from GH therapy. Peripheral edema, myalgias, and arthralgias (especially in the hands and wrists) occur commonly but remit with dosage reduction. Carpal tunnel syndrome can occur. Growth hormone treatment increases the activity of cytochrome P450 isoforms, which may reduce the serum levels of drugs metabolized by that enzyme system (see Chapter 4). There has been no increased incidence of malignancy among patients receiving GH therapy, but such treatment is contraindicated in a patient with a known active malignancy. Proliferative retinopathy may rarely occur. Growth hormone treatment of critically ill patients appears to increase mortality. The long-term health effects of GH treatment in childhood are unknown. The preliminary results from the Safety and Appropriateness of GH in Europe (SAGHE) study are variable. A higher all-cause mortality (mostly due to cardiovascular disease) was found in the GH treatment group in the French arm of the study, but no long-term risks of GH treatment were observed in the study arm from another region of Europe.
MECASERMIN
A small number of children with growth failure have severe IGF-I deficiency that is not responsive to exogenous GH. Causes include mutations in the GH receptor and in the GH receptor signaling pathway, neutralizing antibodies to GH, and IGF-I gene defects. In 2005, the FDA approved two forms of recombinant human IGF-I (rhIGF-I) for treatment of severe IGF-I deficiency that is not responsive to GH: mecasermin and mecasermin rinfabate. Mecasermin is rhIGF-I alone, while mecasermin rinfabate is a complex of rhIGF-I and recombinant human insulin-like growth factor-binding protein-3 (rhIGFBP-3). This binding protein significantly increases the circulating half-life of rhIGF-I. Normally, the great majority of the circulating IGF-I is bound to IGFBP-3, which is produced principally by the liver under the control of GH. Mecasermin rinfabate is not currently available in the United States. Mecasermin is administered subcutaneously twice daily at a recommended starting dosage of 0.04–0.08 mg/kg and increased weekly up to a maximum twice-daily dosage of 0.12 mg/kg.
The most important adverse effect observed with mecasermin is hypoglycemia. To avoid hypoglycemia, the prescribing instructions require consumption of a carbohydrate-containing meal or snack 20 minutes before or after mecasermin administration. Several patients have experienced intracranial hypertension, adenotonsillar hypertrophy, and asymptomatic elevation of liver enzymes.
GROWTH HORMONE ANTAGONISTS
Antagonists of GH are used to reverse the effects of GH-producing cells (somatotrophs) in the anterior pituitary that tend to form GH-secreting tumors. Hormone-secreting pituitary adenomas occur most commonly in adults. In adults, GH-secreting adenomas cause acromegaly, which is characterized by abnormal growth of cartilage and bone tissue, and many organs including skin, muscle, heart, liver, and the gastrointestinal tract. Acromegaly adversely affects the skeletal, muscular, cardiovascular, respiratory, and metabolic systems. When a GH-secreting adenoma occurs before the long bone epiphyses close, it leads to the rare condition, gigantism. Larger pituitary adenomas produce greater amounts of GH and also can impair visual and central nervous system function by encroaching on nearby brain structures. The initial therapy of choice for GH-secreting adenomas is transsphenoidal surgery. Medical therapy with GH antagonists is introduced if GH hypersecretion persists after surgery. These agents include somatostatin analogs and dopamine receptor agonists, which reduce the production of GH, and the novel GH receptor antagonist pegvisomant, which prevents GH from activating GH signaling pathways. Radiation therapy is reserved for patients with inadequate response to surgical and medical therapies.
Somatostatin Analogs
Somatostatin, a 14-amino-acid peptide (Figure 37–2), is found in the hypothalamus, other parts of the central nervous system, the pancreas, and other sites in the gastrointestinal tract. It functions primarily as an inhibitory paracrine factor and inhibits the release of GH, TSH, glucagon, insulin, and gastrin. Somatostatin is rapidly cleared from the circulation, with a half-life of 1–3 minutes. The kidney appears to play an important role in its metabolism and excretion.
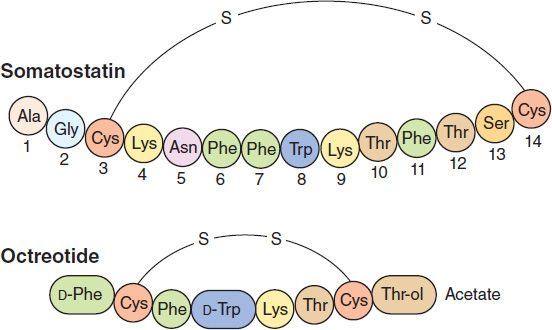
FIGURE 37–2 Above: Amino acid sequence of somatostatin. Below: Sequence of the synthetic analog, octreotide.
Somatostatin has limited therapeutic usefulness because of its short duration of action and multiple effects in many secretory systems. A series of longer-acting somatostatin analogs that retain biologic activity have been developed. Octreotide, the most widely used somatostatin analog (Figure 37–2), is 45 times more potent than somatostatin in inhibiting GH release but only twice as potent in reducing insulin secretion. Because of this relatively reduced effect on pancreatic beta cells, hyperglycemia rarely occurs during treatment. The plasma elimination half-life of octreotide is about 80 minutes, 30 times longer than that of somatostatin.
Octreotide, 50–200 mcg given subcutaneously every 8 hours, reduces symptoms caused by a variety of hormone-secreting tumors: acromegaly, carcinoid syndrome, gastrinoma, glucagonoma, insulinoma, VIPoma, and ACTH-secreting tumor. Other therapeutic use indications include diarrhea—secretory, HIV associated, diabetic, chemotherapy, or radiation induced—and portal hypertension. Somatostatin receptor scintigraphy, using radiolabeled octreotide, is useful in localizing neuroendocrine tumors having somatostatin receptors and helps predict the response to octreotide therapy. Octreotide is also useful for the acute control of bleeding from esophageal varices.
Octreotide acetate injectable long-acting suspension is a slow-release microsphere formulation. It is instituted only after a brief course of shorter-acting octreotide has been demonstrated to be effective and tolerated. Injections into alternate gluteal muscles are repeated at 4-week intervals in doses of 10–40 mg.
Adverse effects of octreotide therapy include nausea, vomiting, abdominal cramps, flatulence, and steatorrhea with bulky bowel movements. Biliary sludge and gallstones may occur after 6 months of use in 20–30% of patients. However, the yearly incidence of symptomatic gallstones is about 1%. Cardiac effects include sinus bradycardia (25%) and conduction disturbances (10%). Pain at the site of injection is common, especially with the long-acting octreotide suspension. Vitamin B12 deficiency may occur with long-term use of octreotide.
A long-acting formulation of lanreotide, another octapeptide somatostatin analog, is approved for treatment of acromegaly. Lanreotide appears to have effects comparable to those of octreotide in reducing GH levels and normalizing IGF-I concentrations.
Pegvisomant
Pegvisomant is a GH receptor antagonist used to treat acromegaly. It is the polyethylene glycol (PEG) derivative of a mutant GH, B2036. Pegylation reduces its clearance and improves its overall clinical effectiveness. Like native GH, pegvisomant has two GH receptor binding sites. However, one of its GH receptor binding sites has increased affinity for the GH receptor, whereas its second GH receptor binding site has reduced affinity. This differential receptor affinity allows the initial step (GH receptor dimerization) but blocks the conformational changes required for signal transduction. In clinical trials, pegvisomant was administered subcutaneously to patients with acromegaly; daily treatment for 12 months or more reduced serum levels of IGF-I into the normal range in 97%. Pegvisomant does not inhibit GH secretion and may lead to increased GH levels and possible adenoma growth. No serious problems have been observed; however, increases in liver enzymes without liver failure have been reported.
THE GONADOTROPINS (FOLLICLE-STIMULATING HORMONE & LUTEINIZING HORMONE) & HUMAN CHORIONIC GONADOTROPIN
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


