CASE STUDY
A healthy 45-year-old physician attending a reunion in a vacation hotel developed dizziness, redness of the skin over the head and chest, and tachycardia while eating. A short time later, another physician at the table developed similar signs and symptoms with marked orthostatic hypotension. The menu included a green salad, sauteed fish with rice, and apple pie. What is the probable diagnosis? How would you treat these patients?
It has long been known that many tissues contain substances that, when released by various stimuli, cause physiologic effects such as reddening of the skin, pain or itching, and bronchospasm. Later, it was discovered that many of these substances are also present in nervous tissue and have multiple functions. Histamine and serotonin (5-hydroxytryptamine, 5-HT) are biologically active amines that function as neurotransmitters and are found in non-neural tissues, have complex physiologic and pathologic effects through multiple receptor subtypes, and are often released locally. Together with endogenous peptides (see Chapter 17), prostaglandins and leukotrienes (see Chapter 18), and cytokines (see Chapter 55), they constitute the autacoid group of drugs.
Because of their broad and largely undesirable peripheral effects, neither histamine nor serotonin has any clinical application in the treatment of disease. However, compounds that selectively activate certain receptor subtypes or selectively antagonize the actions of these amines are of considerable clinical value. This chapter therefore emphasizes the basic pharmacology of the agonist amines and the clinical pharmacology of the more selective agonist and antagonist drugs. The ergot alkaloids, compounds with partial agonist activity at serotonin and several other receptors, are discussed at the end of the chapter.
 HISTAMINE
HISTAMINE
Histamine was synthesized in 1907 and later isolated from mammalian tissues. Early hypotheses concerning the possible physiologic roles of tissue histamine were based on similarities between the effects of intravenously administered histamine and the symptoms of anaphylactic shock and tissue injury. Marked species variation is observed, but in humans histamine is an important mediator of immediate allergic (such as urticaria) and inflammatory reactions, although it plays only a modest role in anaphylaxis. Histamine plays an important role in gastric acid secretion (see Chapter 62) and functions as a neurotransmitter and neuromodulator (see Chapters 6 and 21). Newer evidence indicates that histamine also plays a role in immune functions and chemotaxis of white blood cells.
BASIC PHARMACOLOGY OF HISTAMINE
Chemistry & Pharmacokinetics
Histamine occurs in plants as well as in animal tissues and is a component of some venoms and stinging secretions.
Histamine is formed by decarboxylation of the amino acid L-histidine, a reaction catalyzed in mammalian tissues by the enzyme histidine decarboxylase. Once formed, histamine is either stored or rapidly inactivated. Very little histamine is excreted unchanged. The major metabolic pathways involve conversion to N-methylhistamine, methylimidazoleacetic acid, and imidazoleacetic acid (IAA). Certain neoplasms (systemic mastocytosis, urticaria pigmentosa, gastric carcinoid, and occasionally myelogenous leukemia) are associated with increased numbers of mast cells or basophils and with increased excretion of histamine and its metabolites.
Most tissue histamine is sequestered and bound in granules (vesicles) in mast cells or basophils; the histamine content of many tissues is directly related to their mast cell content. The bound form of histamine is biologically inactive, but as noted below, many stimuli can trigger the release of mast cell histamine, allowing the free amine to exert its actions on surrounding tissues. Mast cells are especially rich at sites of potential tissue injury—nose, mouth, and feet; internal body surfaces; and blood vessels, particularly at pressure points and bifurcations.
Non-mast cell histamine is found in several tissues, including the brain, where it functions as a neurotransmitter. Strong evidence implicates endogenous neurotransmitter histamine in many brain functions such as neuroendocrine control, cardiovascular regulation, thermal and body weight regulation, and sleep and arousal (see Chapter 21).
A second important nonneuronal site of histamine storage and release is the enterochromaffin-like (ECL) cells of the fundus of the stomach. ECL cells release histamine, one of the primary gastric acid secretagogues, to activate the acid-producing parietal cells of the mucosa (see Chapter 62).
Storage & Release of Histamine
The stores of histamine in mast cells can be released through several mechanisms.
A. Immunologic Release
Immunologic processes account for the most important pathophysiologic mechanism of mast cell and basophil histamine release. These cells, if sensitized by IgE antibodies attached to their surface membranes, degranulate explosively when exposed to the appropriate antigen (see Figure 55–5, effector phase). This type of release also requires energy and calcium. Degranulation leads to the simultaneous release of histamine, adenosine triphosphate (ATP), and other mediators that are stored together in the granules. Histamine released by this mechanism is a mediator in immediate (type I) allergic reactions, such as hay fever and acute urticaria. Substances released during IgG- or IgM-mediated immune reactions that activate the complement cascade also release histamine from mast cells and basophils.
By a negative feedback control mechanism mediated by H2 receptors, histamine appears to modulate its own release and that of other mediators from sensitized mast cells in some tissues. In humans, mast cells in skin and basophils show this negative feedback mechanism; lung mast cells do not. Thus, histamine may act to limit the intensity of the allergic reaction in the skin and blood.
Endogenous histamine has a modulating role in a variety of inflammatory and immune responses. Upon injury to a tissue, released histamine causes local vasodilation and leakage of plasma-containing mediators of acute inflammation (complement, C-reactive protein) and antibodies. Histamine has an active chemotactic attraction for inflammatory cells (neutrophils, eosinophils, basophils, monocytes, and lymphocytes). Histamine inhibits the release of lysosome contents and several T- and B-lymphocyte functions. Most of these actions are mediated by H2 or H4 receptors. Release of peptides from nerves in response to inflammation is also probably modulated by histamine acting on presynaptic H3 receptors.
B. Chemical and Mechanical Release
Certain amines, including drugs such as morphine and tubocurarine, can displace histamine from its bound form within cells. This type of release does not require energy and is not associated with mast cell injury or degranulation. Loss of granules from the mast cell also releases histamine, because sodium ions in the extracellular fluid rapidly displace the amine from the complex. Chemical and mechanical mast cell injury causes degranulation and histamine release. Compound 48/80, an experimental drug, selectively releases histamine from tissue mast cells by an exocytotic degranulation process requiring energy and calcium.
Pharmacodynamics
A. Mechanism of Action
Histamine exerts its biologic actions by combining with specific receptors located on the cell membrane. Four different histamine receptors have been characterized and are designated H1–H4; they are described in Table 16–1. Unlike the other amine transmitter receptors discussed previously, no subfamilies have been found within these major types, although different splice variants of several receptor types have been described.
TABLE 16–1 Histamine receptor subtypes.
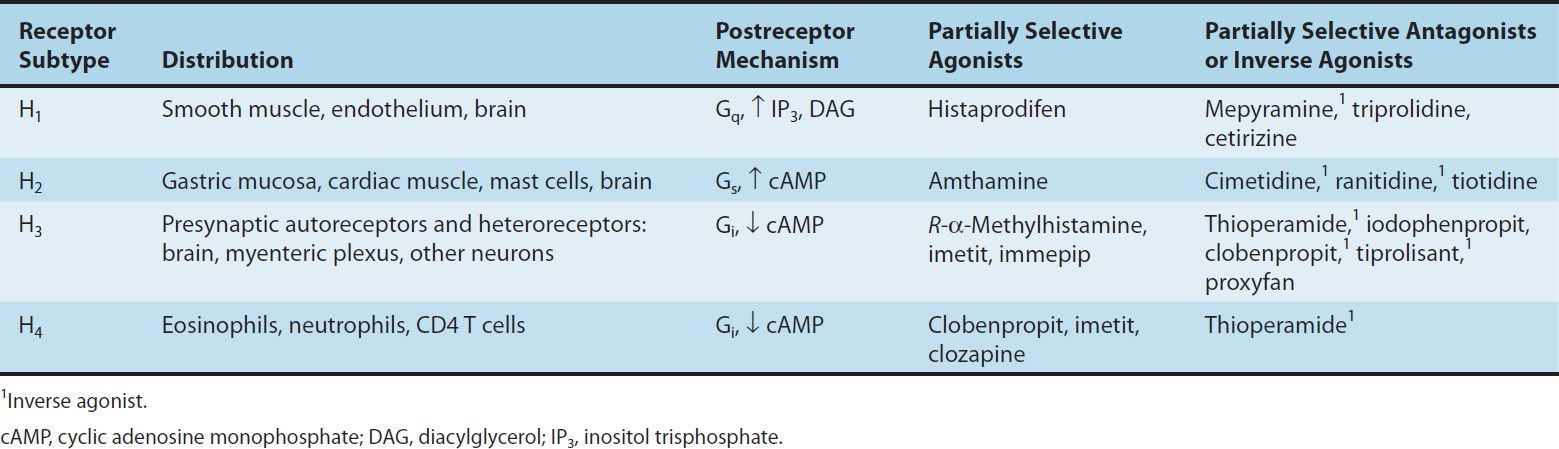
All four receptor types have been cloned and belong to the large superfamily of G-protein-coupled receptors (GPCR). The structures of the H1 and H2 receptors differ significantly and appear to be more closely related to muscarinic and 5-HT1 receptors, respectively, than to each other. The H4 receptor has about 40% homology with the H3 receptor but does not seem to be closely related to any other histamine receptor. All four histamine receptors have been shown to have constitutive activity in some systems; thus, some antihistamines previously considered to be traditional pharmacologic antagonists must now be considered to be inverse agonists (see Chapters 1 and 2). Indeed, many first- and second-generation H1 blockers function as inverse agonists. Furthermore, a single molecule may be an agonist at one histamine receptor and an antagonist or inverse agonist at another. For example, clobenpropit, an agonist at H4 receptors, is an antagonist or inverse agonist at H3 receptors (Table 16–1).
In the brain, H1 and H2 receptors are located on postsynaptic membranes, whereas H3 receptors are predominantly presynaptic. Activation of H1 receptors, which are present in endothelium, smooth muscle cells, and nerve endings, usually elicits an increase in phosphoinositol hydrolysis and an increase in inositol trisphosphate (IP3) and intracellular calcium. Activation of H2 receptors, present in gastric mucosa, cardiac muscle cells, and some immune cells, increases intracellular cyclic adenosine monophosphate (cAMP) via Gs. Like the β2 adrenoceptor, under certain circumstances the H2 receptor may couple to Gq, activating the IP3-DAG (inositol 1,4,5-trisphosphate-diacylglycerol) cascade. Activation of H3 receptors decreases transmitter release from histaminergic and other neurons, probably mediated by a decrease in calcium influx through N-type calcium channels in nerve endings. H4 receptors are found mainly on leukocytes in the bone marrow and circulating blood. H4 receptors appear to have very important chemotactic effects on eosinophils and mast cells. In this role, they seem to play a part in inflammation and allergy. They may also modulate production of these cell types and they may mediate, in part, the previously recognized effects of histamine on cytokine production.
B. Tissue and Organ System Effects of Histamine
Histamine exerts powerful effects on smooth and cardiac muscle, on certain endothelial and nerve cells, on the secretory cells of the stomach, and on inflammatory cells. However, sensitivity to histamine varies greatly among species. Guinea pigs are exquisitely sensitive; humans, dogs, and cats somewhat less so; and mice and rats very much less so.
1. Nervous system—Histamine is a powerful stimulant of sensory nerve endings, especially those mediating pain and itching. This H1-mediated effect is an important component of the urticarial response and reactions to insect and nettle stings. Some evidence suggests that local high concentrations can also depolarize efferent (axonal) nerve endings (see Triple Response, item 8 in this list). In the mouse, and probably in humans, respiratory neurons signaling inspiration and expiration are modulated by H1 receptors. H1 and H3 receptors play important roles in appetite and satiety; antipsychotic drugs that block these receptors cause significant weight gain (see Chapter 29). These receptors may also participate in nociception. Presynaptic H3 receptors play important roles in modulating release of several transmitters in the nervous system. H3 agonists reduce the release of acetylcholine, amine, and peptide transmitters in various areas of the brain and in peripheral nerves. An investigational inverse H3 agonist, pitolisant (BF2649), appears to reduce drowsiness in patients with narcolepsy.
2. Cardiovascular system—In humans, injection or infusion of histamine causes a decrease in systolic and diastolic blood pressure and an increase in heart rate. The blood pressure changes are caused by the direct vasodilator action of histamine on arterioles and precapillary sphincters; the increase in heart rate involves both stimulatory actions of histamine on the heart and a reflex tachycardia. Flushing, a sense of warmth, and headache may also occur during histamine administration, consistent with the vasodilation. Vasodilation elicited by small doses of histamine is caused by H1-receptor activation and is mediated mainly by release of nitric oxide from the endothelium (see Chapter 19). The decrease in blood pressure is usually accompanied by a reflex tachycardia. Higher doses of histamine activate the H2-mediated cAMP process of vasodilation and direct cardiac stimulation. In humans, the cardiovascular effects of small doses of histamine can usually be antagonized by H1-receptor antagonists alone.
Histamine-induced edema results from the action of the amine on H1 receptors in the vessels of the microcirculation, especially the postcapillary vessels. The effect is associated with the separation of the endothelial cells, which permits the transudation of fluid and molecules as large as small proteins into the perivascular tissue. This effect is responsible for urticaria (hives), which signals the release of histamine in the skin. Studies of endothelial cells suggest that actin and myosin within these cells cause contraction, resulting in separation of the endothelial cells and increased permeability.
Direct cardiac effects of histamine include both increased contractility and increased pacemaker rate. These effects are mediated chiefly by H2 receptors. In human atrial muscle, histamine can also decrease contractility; this effect is mediated by H1 receptors. The physiologic significance of these cardiac actions is not clear. Some of the cardiovascular signs and symptoms of anaphylaxis are due to released histamine, although several other mediators are involved and are much more important than histamine in humans.
3. Bronchiolar smooth muscle—In both humans and guinea pigs, histamine causes bronchoconstriction mediated by H1 receptors. In the guinea pig, this effect is the cause of death from histamine toxicity, but in humans with normal airways, bronchoconstriction following small doses of histamine is not marked. However, patients with asthma are very sensitive to histamine. The bronchoconstriction induced in these patients probably represents a hyperactive neural response, since such patients also respond excessively to many other stimuli, and the response to histamine can be blocked by autonomic blocking drugs such as ganglion blocking agents as well as by H1-receptor antagonists (see Chapter 20). Although methacholine provocation is more commonly used, tests using small doses of inhaled histamine have been used in the diagnosis of bronchial hyperreactivity in patients with suspected asthma or cystic fibrosis. Such individuals may be 100 to 1000 times more sensitive to histamine (and methacholine) than are normal subjects. Curiously, a few species (eg, rabbit) respond to histamine with bronchodilation, reflecting the dominance of the H2 receptor in their airways.
4. Gastrointestinal tract smooth muscle—Histamine causes contraction of intestinal smooth muscle, and histamine-induced contraction of guinea pig ileum is a standard bioassay for this amine. The human gut is not as sensitive as that of the guinea pig, but large doses of histamine may cause diarrhea, partly as a result of this effect. This action of histamine is mediated by H1 receptors.
5. Other smooth muscle organs—In humans, histamine generally has insignificant effects on the smooth muscle of the eye and genitourinary tract. However, pregnant women suffering anaphylactic reactions may abort as a result of histamine-induced contractions, and in some species the sensitivity of the uterus is sufficient to form the basis for a bioassay.
6. Secretory tissue—Histamine has long been recognized as a powerful stimulant of gastric acid secretion and, to a lesser extent, of gastric pepsin and intrinsic factor production. The effect is caused by activation of H2 receptors on gastric parietal cells and is associated with increased adenylyl cyclase activity, cAMP concentration, and intracellular Ca2+ concentration. Other stimulants of gastric acid secretion such as acetylcholine and gastrin do not increase cAMP even though their maximal effects on acid output can be reduced—but not abolished—by H2-receptor antagonists. These actions are discussed in more detail in Chapter 62. Histamine also stimulates secretion in the small and large intestine. In contrast, H3-selective histamine agonists inhibit acid secretion stimulated by food or pentagastrin in several species.
Histamine has much smaller effects on the activity of other glandular tissue at ordinary concentrations. Very high concentrations can cause catecholamine release from the adrenal medulla.
7. Metabolic effects—Recent studies of H3-receptor knockout mice demonstrate that absence of this receptor results in increased food intake, decreased energy expenditure, and obesity. They also show insulin resistance and increased blood levels of leptin and insulin. It is not yet known whether the H3 receptor has a similar role in humans, but research is underway to determine whether H3 agonists are useful in the treatment of obesity.
8. The “triple response”—Intradermal injection of histamine causes a characteristic red spot, edema, and flare response. The effect involves three separate cell types: smooth muscle in the microcirculation, capillary or venular endothelium, and sensory nerve endings. At the site of injection, a reddening appears owing to dilation of small vessels, followed soon by an edematous wheal at the injection site and a red irregular flare surrounding the wheal. The flare is said to be caused by an axon reflex. A sensation of itching may accompany these effects.
Similar local effects may be produced by injecting histamine liberators (compound 48/80, morphine, etc) intradermally or by applying the appropriate antigens to the skin of a sensitized person. Although most of these local effects can be prevented by adequate doses of an H1-receptor-blocking agent, H2 and H3 receptors may also be involved.
9. Other effects possibly mediated by histamine receptors—In addition to the local stimulation of peripheral pain nerve endings via H3 and H1 receptors, histamine may play a role in nociception in the central nervous system. Burimamide, an early candidate for H2-blocking action, and newer analogs with no notable effect on H1, H2, or H3 receptors, have been shown to have significant analgesic action in rodents when administered into the central nervous system. The analgesia is said to be comparable to that produced by opioids, but tolerance, respiratory depression, and constipation have not been reported.
Other Histamine Agonists
Small substitutions on the imidazole ring of histamine significantly modify the selectivity of the compounds for the histamine receptor subtypes. Some of these are listed in Table 16–1.
CLINICAL PHARMACOLOGY OF HISTAMINE
Clinical Uses
In pulmonary function laboratories, histamine aerosol has been used as a provocative test of bronchial hyperreactivity. Histamine has no other current clinical applications.
Toxicity & Contraindications
Adverse effects of histamine release, like those following administration of histamine, are dose related. Flushing, hypotension, tachycardia, headache, urticaria, bronchoconstriction, and gastrointestinal upset are noted. These effects are also observed after the ingestion of spoiled fish (scombroid fish poisoning), and histamine produced by bacterial action in the flesh of improperly stored fish is the major causative agent.
Histamine should not be given to patients with asthma (except as part of a carefully monitored test of pulmonary function) or to patients with active ulcer disease or gastrointestinal bleeding.
HISTAMINE ANTAGONISTS
The effects of histamine released in the body can be reduced in several ways. Physiologic antagonists, especially epinephrine, have smooth muscle actions opposite to those of histamine, but they act at different receptors. This is important clinically because injection of epinephrine can be lifesaving in systemic anaphylaxis and in other conditions in which massive release of histamine—and other more important mediators—occurs.
Release inhibitors reduce the degranulation of mast cells that results from immunologic triggering by antigen-IgE interaction. Cromolyn and nedocromil appear to have this effect (see Chapter 20) and have been used in the treatment of asthma. Beta2-adrenoceptor agonists also appear capable of reducing histamine release.
Histamine receptor antagonists represent a third approach to the reduction of histamine-mediated responses. For over 60 years, compounds have been available that competitively antagonize many of the actions of histamine on smooth muscle. However, not until the H2-receptor antagonist burimamide was described in 1972 was it possible to antagonize the gastric acid-stimulating activity of histamine. The development of selective H2-receptor antagonists has led to more effective therapy for peptic disease (see Chapter 62). Selective H3 and H4 antagonists are not yet available for clinical use. However, potent and partially selective experimental H3-receptor antagonists, thioperamide and clobenpropit, have been developed.
 HISTAMINE RECEPTOR ANTAGONISTS
HISTAMINE RECEPTOR ANTAGONISTS
H1-RECEPTOR ANTAGONISTS
Compounds that competitively block histamine or act as inverse agonists at H1 receptors have been used in the treatment of allergic conditions for many years, and in the discussion that follows are referred to as antagonists. Many H1 antagonists are currently marketed in the USA. A large number are available without prescription, both alone and in combination formulations such as “cold pills” and “sleep aids” (see Chapter 63).
BASIC PHARMACOLOGY OF H1-RECEPTOR ANTAGONISTS
Chemistry & Pharmacokinetics
The H1 antagonists are conveniently divided into first-generation and second-generation agents. These groups are distinguished by the relatively strong sedative effects of most of the first-generation drugs. The first-generation agents are also more likely to block autonomic receptors. Second-generation H1 blockers are less sedating, owing in part to reduced distribution into the central nervous system. All the H1 antagonists are stable amines with the general structure illustrated in Figure 16–1. Doses of some of these drugs are given in Table 16–2.
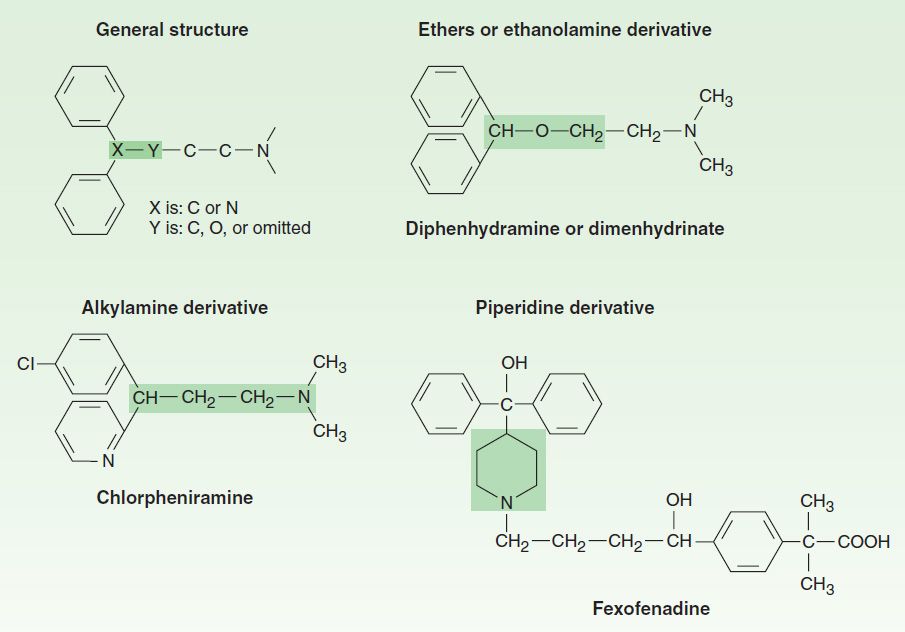
FIGURE 16–1 General structure of H1-antagonist drugs and examples of the major subgroups. Subgroup names are based on the shaded moieties.
TABLE 16–2 Some H1 antihistaminic drugs in clinical use.
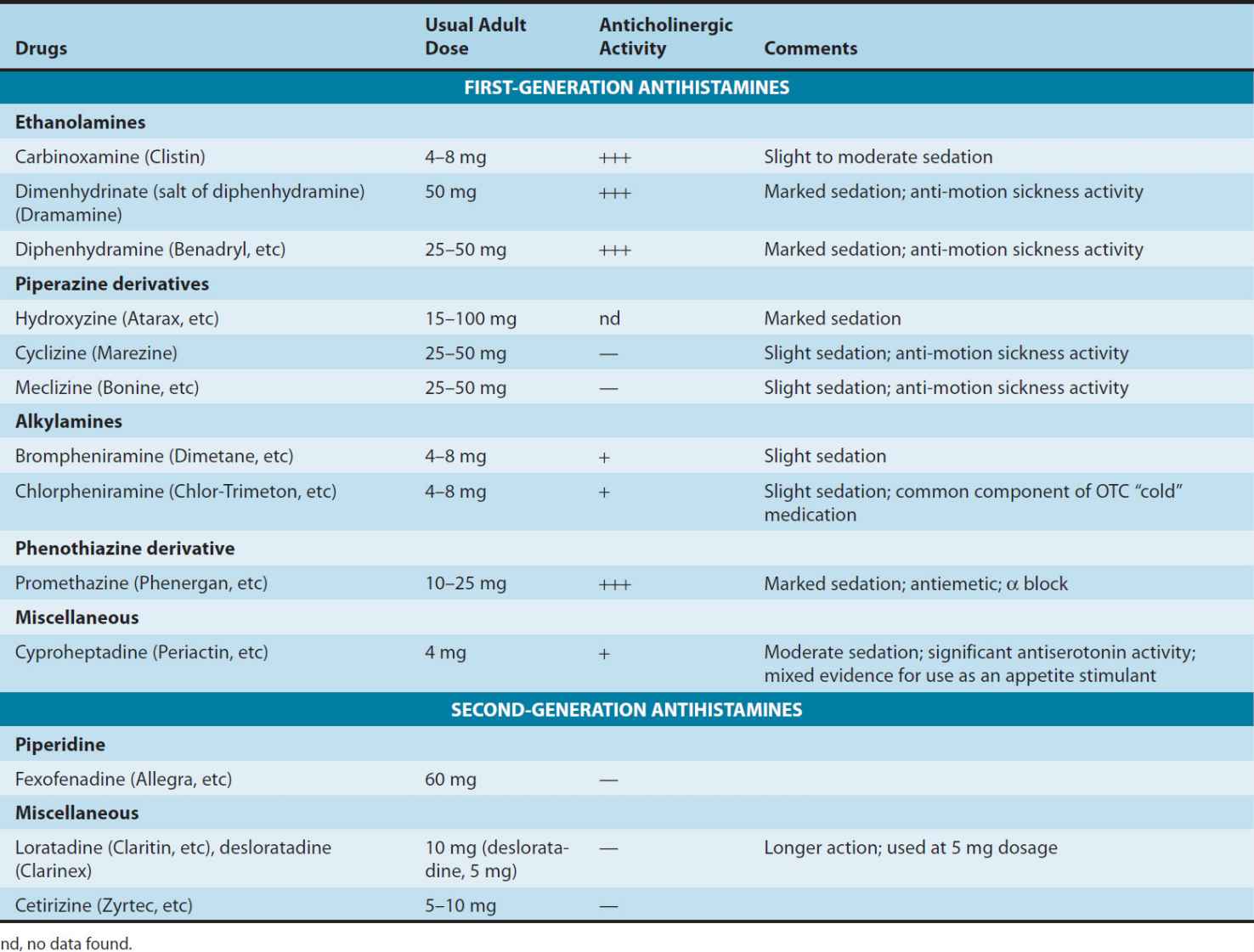
These agents are rapidly absorbed after oral administration, with peak blood concentrations occurring in 1–2 hours. They are widely distributed throughout the body, and the first-generation drugs enter the central nervous system readily. Some of them are extensively metabolized, primarily by microsomal systems in the liver. Several of the second-generation agents are metabolized by the CYP3A4 system and thus are subject to important interactions when other drugs (such as ketoconazole) inhibit this subtype of P450 enzymes. Most of the drugs have an effective duration of action of 4–6 hours following a single dose, but meclizine and several second-generation agents are longer-acting, with a duration of action of 12–24 hours. The newer agents are considerably less lipid-soluble than the first-generation drugs and are substrates of the P-glycoprotein transporter in the blood-brain barrier; as a result they enter the central nervous system with difficulty or not at all. Many H1 antagonists have active metabolites. The active metabolites of hydroxyzine, terfenadine, and loratadine are available as drugs (cetirizine, fexofenadine, and desloratadine, respectively).
Pharmacodynamics
Both neutral H1 antagonists and inverse H1 agonists reduce or block the actions of histamine by reversible competitive binding to the H1 receptor. Several have been clearly shown to be inverse agonists, and it is possible that all act by this mechanism. They have negligible potency at the H2 receptor and little at the H3 receptor. For example, histamine-induced contraction of bronchiolar or gastrointestinal smooth muscle can be completely blocked by these agents, but histamine-stimulated gastric acid secretion and the stimulation of the heart are unaffected.
The first-generation H1-receptor antagonists have many actions in addition to blockade of the actions of histamine. The large number of these actions probably results from the similarity of the general structure (Figure 16–1) to the structure of drugs that have effects at muscarinic cholinoceptor, a adrenoceptor, serotonin, and local anesthetic receptor sites. Some of these actions are of therapeutic value and some are undesirable.
1. Sedation—A common effect of first-generation H1 antagonists is sedation, but the intensity of this effect varies among chemical subgroups (Table 16–2) and among patients as well. The effect is sufficiently prominent with some agents to make them useful as “sleep aids” (see Chapter 63) and unsuitable for daytime use. The effect resembles that of some antimuscarinic drugs and is considered very different from the disinhibited sedation produced by sedative-hypnotic drugs. Compulsive use has not been reported. At ordinary dosages, children occasionally (and adults rarely) manifest excitation rather than sedation. At very high toxic dose levels, marked stimulation, agitation, and even seizures may precede coma. Second-generation H1 antagonists have little or no sedative or stimulant actions. These drugs (or their active metabolites) also have far fewer autonomic effects than the first-generation antihistamines.
2. Antinausea and antiemetic actions—Several first-generation H1 antagonists have significant activity in preventing motion sickness (Table 16–2). They are less effective against an episode of motion sickness already present. Certain H1 antagonists, notably doxylamine (in Bendectin), were used widely in the past in the treatment of nausea and vomiting of pregnancy (see below). Although Bendectin was withdrawn in 1983, a similar formulation, combining doxylamine and pyridoxine (Diclegis), was approved by the FDA in 2013.
3. Antiparkinsonism effects—Some of the H1 antagonists, especially diphenhydramine, have significant acute suppressant effects on the extrapyramidal symptoms associated with certain antipsychotic drugs. This drug is given parenterally for acute dystonic reactions to antipsychotics.
4. Antimuscarinic actions—Many first-generation agents, especially those of the ethanolamine and ethylenediamine subgroups, have significant atropine-like effects on peripheral muscarinic receptors. This action may be responsible for some of the (uncertain) benefits reported for nonallergic rhinorrhea but may also cause urinary retention and blurred vision.
5. Adrenoceptor-blocking actions—Alpha-receptor-blocking effects can be demonstrated for many H1 antagonists, especially those in the phenothiazine subgroup, eg, promethazine. This action may cause orthostatic hypotension in susceptible individuals. Beta-receptor blockade is not significant.
6. Serotonin-blocking actions—Strong blocking effects at serotonin receptors have been demonstrated for some first-generation H1 antagonists, notably cyproheptadine. This drug is promoted as an antiserotonin agent and is discussed with that drug group. Nevertheless, its structure resembles that of the phenothiazine antihistamines, and it is a potent H1-blocking agent.
7. Local anesthesia—Several first-generation H1 antagonists are potent local anesthetics. They block sodium channels in excitable membranes in the same fashion as procaine and lidocaine. Diphenhydramine and promethazine are actually more potent than procaine as local anesthetics. They are occasionally used to produce local anesthesia in patients allergic to conventional local anesthetic drugs. A small number of these agents also block potassium channels; this action is discussed below (see Toxicity).
8. Other actions—Certain H1 antagonists, eg, cetirizine, inhibit mast cell release of histamine and some other mediators of inflammation. This action is not due to H1-receptor blockade and may reflect an H4-receptor effect (see below). The mechanism is not fully understood but could play a role in the beneficial effects of these drugs in the treatment of allergies such as rhinitis. A few H1 antagonists (eg, terfenadine, acrivastine) have been shown to inhibit the P-glycoprotein transporter found in cancer cells, the epithelium of the gut, and the capillaries of the brain. The significance of this effect is not known.
CLINICAL PHARMACOLOGY OF H1-RECEPTOR ANTAGONISTS
Clinical Uses
First-generation H1-receptor blockers are still commonly used over-the-counter drugs. The prevalence of allergic conditions and the relative safety of the drugs contribute to this heavy use. However, the fact that they do cause sedation contributes to heavy prescribing as well as over-the-counter use of second-generation antihistamines.
A. Allergic Reactions
The H1 antihistaminic agents are often the first drugs used to prevent or treat the symptoms of allergic reactions. In allergic rhinitis (hay fever), the H1 antagonists are second-line drugs after glucocorticoids administered by nasal spray. In urticaria, in which histamine is the primary mediator, the H1 antagonists are the drugs of choice and are often quite effective if given before exposure. However, in bronchial asthma, which involves several mediators, the H1 antagonists are largely ineffective.
Angioedema may be precipitated by histamine release but appears to be maintained by peptide kinins that are not affected by antihistaminic agents. For atopic dermatitis, antihistaminic drugs such as diphenhydramine are used mostly for their sedative side effect, which reduces awareness of itching.
The H1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


