Amsterdam Criteria II
There should be at least three relatives with an HNPCC-associated cancer (CRC, cancer of the endometrium, small bowel, ureter, or renal pelvis)
One should be a first-degree relative of the other two
At least two successive generations should be affected
At least one should be diagnosed before age 50 years
Familial adenomatous polyposis should be excluded in the CRC case(s), if any
Tumors should be verified by pathological examination
Revised Bethesda Guidelines
CRC in a patient who is less than 50 years of age
Presence of synchronous, metachronous colorectal, or other HNPCC-associated tumors,a regardless of age
CRC with the MSI-H histologyb diagnosed in a patient who is less than 60 years of age
CRC diagnosed in one or more first-degree relatives with an HNPCC-related tumor, with one of the cancers being diagnosed under age 50 years
CRC diagnosed in two or more first- or second-degree relatives with HNPCC-related tumors, regardless of age
CRC arising in the setting of defective MMR has a tendency toward a number of pathologic features, including location in the proximal colon, mucinous or signet ring differentiation, poor differentiation, and prominent lymphocytic inflammation in and around the tumor. Examples of histologic features of MMR-deficient tumors are shown in Fig. 24.1. Recognition of histologic patterns has been shown to be highly sensitive for tumors with defective MMR, with reported sensitivity in some studies greater than 90 %. The specificity of histology, however, is only 55–67 %, making additional testing a necessary component of tumor evaluation [14–16].
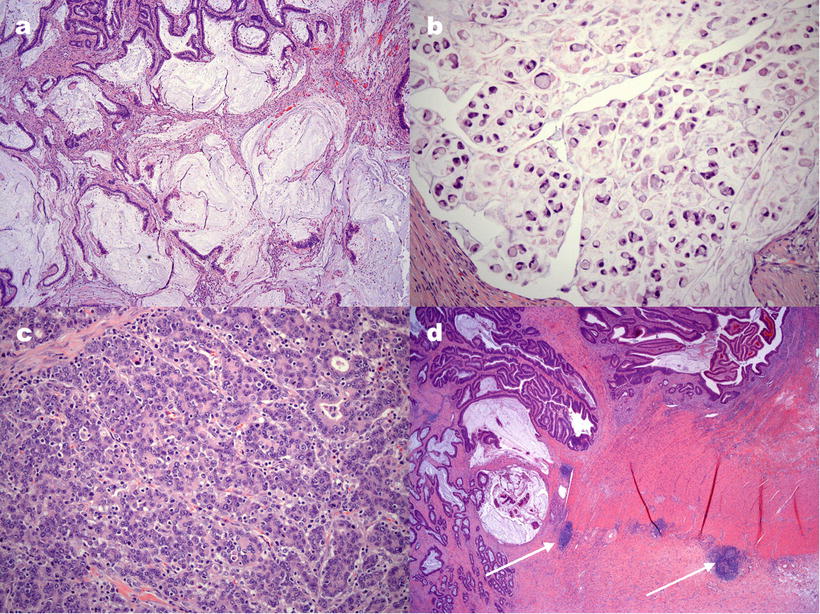
Figure 24.1
Examples of histological features of colorectal carcinomas with defective DNA mismatch repair. (a) Extracellular mucin pools in a mucinous adenocarcinoma. (b) Signet ring cell adenocarcinoma with tumor cells containing a single, large mucin vacuole. (c) Tumor-infiltrating lymphocytes are seen as small, round cells, often with a surrounding clear “halo,” infiltrating among tumor cell glands or aggregates. (d) Crohn’s-like lymphocytic response is illustrated by discrete lymphocytic aggregates (arrows) away from the tumor, resembling the transmural inflammation of Crohn’s disease
Lynch syndrome is associated with several other cancers (listed among the Bethesda Guidelines). The association of some of these cancers with CRC has been noted by medical researchers over the years, and these have been given eponymous syndromes such as Turcot (glioblastoma) and Muir–Torre (keratoacanthoma and sebaceous tumors) syndromes. The importance of gynecologic cancers in the disease burden cannot be stressed enough, with uterine and ovarian cancer comprising half of the presenting cancers in women with Lynch syndrome [17]. The overall risk of CRC by age 70 in Lynch carriers has been reported as high as 82 %, although some studies have shown a lower risk of 30–74 %, with men at significantly higher risk than women [18–20]. The risk of uterine cancer in women with Lynch syndrome is 34–60 % [18, 20, 21]. For some less common tumors such as gastric, small bowel, and urinary tract cancers, the lifetime risk of cancer is approximately 5–15 % [22].
Molecular Basis of Disease
In 1993, through the work of multiple groups working independently, the molecular basis for Lynch syndrome began to be uncovered. This was first recognized in HNPCC tumors as changes in the size of microsatellites, tandem repetitive sequences of DNA which make up about 3 % of the human genome. The repeating sequence in these microsatellites ranges in size from 1 to 13 bases, although microsatellites longer than 5 bases are not typically used in testing. In HNPCC tumors, the number of repeats in microsatellites expanded or contracted, resulting in a shift in the size of the microsatellites. These changes were called replication errors or microsatellite instability (originally abbreviated MIN but now usually shown as MSI). MSI was found to be much more common in tumors of families with HNPCC than in sporadic CRC [23–25].
Soon after the discovery of MSI in HNPCC, the phenomenon was linked to defective function of proteins involved in the repair of errors introduced during DNA replication. These proteins have homologs, MutS and MutL, originally described in E. coli, whose function is to recognize and correct DNA heteroduplexes of mismatched base pairs [26]. Microsatellites, due to the repetitive nature of their sequences, appear to represent a “slippery” template along which DNA polymerase is prone to skip forward or backward during replication, creating small loops of unpaired bases in the process [27]. Depending on the direction of the slippage, the newly synthesized strand will contain a greater or lesser number of repeats than the template. Single base–base mismatches can occur when an incorrect nucleotide is incorporated into the new strand. An intact MMR system recognizes these alterations and directs a multistep process of strand breakage, excision of the error, and resynthesis of the strand, thereby resulting in a corrected sequence. The connection between the MSI and the MMR system was first shown by the identification of germline mutations in MSH2, the human homolog of mutS, in families with Lynch syndrome, and later in the MutL homolog MLH1 [28, 29]. Most of these mutations result in a premature stop codon and a truncated protein. Less commonly, missense mutations affect protein-protein interactions that are essential for MMR function.
CRC arising in Lynch syndrome are believed to follow a typical adenoma–carcinoma sequence in common with non-Lynch tumors. This has been shown through the presence of adenomas adjacent to early carcinomas identified in prophylactic colectomies performed in Lynch syndrome carriers [30]. Furthermore, adenomas arising in Lynch syndrome, particularly those that show high histologic grade, often have evidence of MSI prior to the development of invasive carcinoma [31]. While there have been reports of MSI in histologically normal cells and tissues of patients with Lynch syndrome, in most cases, testing of normal tissue in Lynch syndrome carriers does not demonstrate MSI, and the presence of MMR proteins can be demonstrated with immunohistochemical staining [32–35]. Thus, despite the loss of one functional copy of a MMR gene, DNA MMR largely remains intact until a second genetic hit results in deletion or mutation of the functioning allele.
At least seven MMR proteins involved in DNA repair during mitosis have been identified in humans [36]. Four of these, MLH1, PMS2, MSH2, and MSH6, appear to be largely responsible for Lynch syndrome, with mutations in MLH1 and MSH2 causing approximately 90 % of cases [37]. The MMR proteins function as heterodimers, and MLH1 and MSH2 serve as the obligate components of dimers homologous to the bacterial mutL and mutS, respectively. MLH1 primarily pairs with PMS2 but can also be paired with PMS1 or MSH3. MSH2 pairs primarily with MSH6, and MSH3 is a secondary partner [36]. The specific combinations formed determine the types of DNA mismatches that are corrected. For example, MSH2 and MSH6 can repair single base pair mismatches as well as longer insertions and deletions, while MSH2 and MSH3 mainly correct larger mismatches [26]. The different pairings and their functions seem to have clinical relevance, as shown by the fact that different Lynch mutations do not appear to be equal with respect to the clinical manifestations of the syndrome and the associated cancer risk. Carriers with mutations in the obligate components MLH1 and MSH2, without which MMR dimers cannot function, have an approximately four-fold higher risk of CRC than do those with an MSH6 mutation [21]. This may be explained by substitution of MSH6 by MSH3 in the MutS-type dimer, partially compensating for MSH6 loss. The effect of this substitution is restoration of repair of longer mutations, while single base errors may go uncorrected [38].
In addition to HNPCC-associated cancers, sporadic CRC (i.e., those not associated with a strong family history of CRC) also can have MSI [39]. These microsatellite-unstable tumors make up approximately 15–25 % of sporadic CRC, a much greater number of tumors than those attributable to Lynch syndrome [40]. Like Lynch-associated CRC, sporadic tumors with MSI have a tendency toward a right-sided proximal location in the colon, but the age of onset of these tumors does not differ significantly from MMR-proficient tumors [41, 42]. The precursor lesion of these cancers, sessile serrated adenoma, lacks the cellular atypia of tubular adenomas that precede Lynch-associated tumors and has the saw-toothed microscopic appearance of a common hyperplastic polyp. However, the sessile serrated adenoma differs from hyperplastic polyps by increased architectural complexity, which is due to altered distribution of proliferating cells in colonic glands [43]. Analysis of MMR genes showed that most of the sporadic tumors lack either germline or somatic mutations in MMR genes, yet immunohistochemical staining for MMR proteins shows that these tumors commonly lack expression of MLH1 [44]. This apparent discrepancy was explained by the recognition that the MLH1 promoter was epigenetically silenced via hypermethylation in sporadic CRC with MSI [45]. Further, MLH1 hypermethylation is closely linked to the activating V600E mutation in the BRAF kinase, with BRAF mutation seen in 31–87 % of CRC with silenced MLH1. Significantly for diagnostic purposes, BRAF mutation is virtually never seen in Lynch syndrome-associated CRC [46–48]. A causal relationship between BRAF mutation and MLH1 silencing has yet to be discovered. MLH1 hypermethylation also has been demonstrated in sporadic endometrial cancers with MSI. However, in stark contrast with what is seen in the colon, these tumors consistently lack BRAF mutation, suggesting that MLH1 hypermethylation can occur independently of BRAF mutation [49]. Thus, BRAF mutation may indicate an MLH1-methylated CRC but may not be the underlying etiology. Rarely, constitutional MLH1 methylation may be inherited, resulting in Lynch syndrome without protein-coding mutations in MMR genes. While this constitutes <1% of all Lynch syndrome patients, it is an important Lynch etiology in individuals who are MLH1-deficient but negative for MLH1 mutation [50].
While DNA replication errors in microsatellites can be used as an indicator of defective mismatch repair function, it is not clear what role MSI may play in vivo in cancer pathogenesis. The Human Genome Project has revealed that many microsatellites are located in the coding regions of hundreds of genes. These include genes encoding proteins with important roles in signal transduction (TGFBII and IGFIIR, receptors for TGFB and insulin-like growth factor, respectively), tumor suppression (PTEN), and apoptosis (BAX). Ironically, the genes for two of the MMR proteins, MSH3 and MSH6, also contain microsatellites. Supporting the idea that instability in these and other microsatellite-containing genes can affect colon carcinogenesis is the observation that frameshift mutations in these genes have been identified in tumors from HNPCC families, and this instability can occur as early as the adenoma stage [51, 52].
Clinical Utility of Testing
The primary goal of most testing for defective mismatch repair is to screen for Lynch syndrome carriers. The high risk of malignancy in this population makes their identification, increased surveillance, and consideration of medical and surgical prophylaxis of critical importance in preventing morbidity and mortality. Carriers of MMR protein mutations do not have an increased rate of development of precancerous adenomas, but progression from adenoma to carcinoma appears to be accelerated [53]. For this reason, the recommended interval for colonoscopy in this population is every 1–2 years, shorter than the 5-year interval for non-carriers. Increased frequency of colonoscopy in surveillance appears to result in fewer invasive cancers due to removal at the adenoma stage [54, 55].
In addition to increased surveillance, another strategy under investigation to protect against cancer is chemoprevention. Aspirin has been used in a long-term trial (CAPP2) with Lynch carriers to see if it lowers the incidence of CRC. After nearly 5-year average follow-up, participants taking aspirin had a nearly 40 % reduction in CRC risk compared to those taking placebo [56]. The most definitive preventive measure is surgical resection of organs at high risk of malignancy, including subtotal or greater colectomy for Lynch carriers undergoing CRC resection. This is a complex issue that is beyond the scope of this chapter, but it has been addressed by a number of authors and been shown to be both medically effective and cost effective [55, 57, 58].
Once one Lynch carrier has been identified, the specific germline MMR gene mutation detected can be assayed in the patient’s blood relatives, greatly expanding the importance of the initial detection. The first-degree relatives of a Lynch carrier each have a 50 % risk of being a carrier. Given the potential interventions for Lynch syndrome carriers, some have advocated for universal screening of all CRC for evidence of defective MMR [59, 60]. While the specific recommendations for screening method differ, universal screening may be cost effective [13, 61, 62].
Determination of MMR status has prognostic and predictive significance. Clinical studies of patients with MMR-deficient CRC have shown that these tumors present with more advanced local disease (have deeper invasion into the colonic wall), yet paradoxically, patients with these tumors have a longer survival than those with MMR-proficient cancer [63–65]. A very thorough review of survival studies by Popat et al. estimated the survival advantage of patients with MMR-deficient CRC to be 15 % or more [66]. Although both sporadic and Lynch-associated MMR-deficient tumors have a better prognosis, this effect is most pronounced in patients with Lynch syndrome [63]. It is unlikely that the younger age of presentation of patients with Lynch syndrome completely explains the improved survival. When early-onset CRC patients were compared with respect to MMR function, there remained a trend toward improved survival in those with defective MMR [67]. For reasons that are not clear, MMR-deficient tumors show less aggressive behavior in terms of lymphatic and distant metastasis and prolonged time to recurrence [63, 68, 69].
The improved survival in patients with MMR-deficient tumors means there is less opportunity for benefit from chemotherapy. Patients and their physicians may feel that the adverse effects of chemotherapy outweigh the modest improvement in survival (5 % at 5 years in stage II patients) that has been associated with adjuvant treatment [70]. Additionally, in patients with stage II and III disease, studies have shown that while fluorouracil treatment of tumors with intact MMR improves survival, patients with MMR-deficient tumors may not benefit [71, 72].
Available Assays
The majority of testing is directed toward identifying tumors with defective MMR, both as a means of screening for potential Lynch carriers and for its prognostic and therapeutic implications. Once potential carriers have been identified, additional testing can identify the presence of a germline mutation in an MMR protein to confirm a Lynch syndrome diagnosis and to allow counseling and targeted testing of family members for carrier status. However, since most MMR-deficient tumors arise sporadically, the number of patients who require germline mutation testing is relatively small, and the focus of this section is on screening assays. Protocols for germline testing have been published and involve both direct sequencing for small mutations and denaturing high-pressure liquid chromatography for large rearrangements [73]. Hundreds of Lynch syndrome mutations have been identified and are catalogued online by the International Society for Gastrointestinal Hereditary Tumours (InSiGHT; http://www.insight-group.org) [74]. Different Lynch syndrome mutations have different cancer risks, with MLH1 and MSH2 mutations associated with approximately four-fold greater risk of cancer than MSH6 mutation [21]. Thus, identification of the specific mutation can provide useful information for counseling carriers and their family members.
Immunohistochemistry
The most readily available assay for detecting MMR deficiency is immunohistochemical (IHC) staining of tissue sections for MMR proteins. This can be performed by most histology laboratories using standard protocols with commercial antibodies against MLH1, PMS2, MSH2, and MSH6. This technique directly detects the presence or absence of these MMR proteins in the tumor but does not assay for the activity of the MMR proteins. MMR mutations result in a truncated or otherwise nonfunctional protein that is then degraded by the cell [75, 76]. IHC, then, identifies the loss of one or more mismatch repair proteins in a tumor.
Related to their function to ensure the fidelity of DNA replication, MMR proteins are normally found in the nucleus. In a tumor with a functioning MMR system, staining for each MMR protein will reveal positive staining in the nuclei of tumor cells. This system is also at work in non-tumor cells, which also should have positive nuclear staining. Evaluation of non-tumor cells can serve as an internal positive control for each stain [77]. Although every cell in a MMR-proficient tumor is expected to be positive if the MMR proteins are intact, in practice, this is usually not the case. As is seen with many IHC stains, staining often is variable throughout the tumor. Indeed, non-tumor cells may show the same inconsistency in staining [78]. It is unclear if this represents variable fixation/antigen retrieval issues which abound with IHC, biologic variability at the cell level, or a combination of these factors. In general, MMR proteins that are nonfunctional will fail to dimerize and will be degraded. Using IHC, this typically corresponds to widespread loss of nuclear staining in the tumor cells [77]. Similarly, epigenetic silencing of MLH1 expression also results in a loss of staining [44]. An example of loss of MMR staining by IHC is shown in Fig. 24.2.
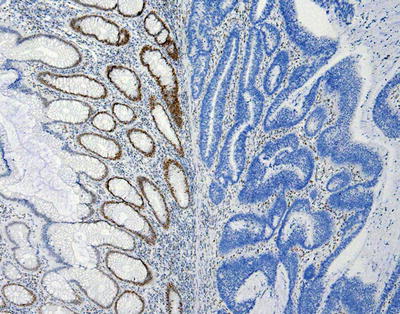
Figure 24.2
Example of loss of immunohistochemical staining for the mismatch repair protein PMS2 in a colon carcinoma. The normal glands on the left show positive, brown staining for PMS2 in cell nuclei, while the cancerous glands on the right lack the brown staining. The blue staining present in the nucleus is a counterstain to show the tissues. Note that even in the normal tissue on the left, not every cell will stain positively
MMR protein heterodimer composition allows prediction of the underlying molecular defect based on the pattern of protein loss. Usually, when the obligatory partner of a MMR component is lost, that protein will be lost as well. On the other hand, MMR proteins that may dimerize with multiple partners can still be present if one of the partners is deficient. For example, PMS2 can only pair with MLH1, and therefore, loss of MLH1 most often results in lack of staining for both MLH1 and PMS2. MLH1, on the other hand, can pair with proteins other than PMS2, and loss of PMS2 may occur without MLH1 loss. Table 24.2 demonstrates the common protein loss patterns and the causative genetic defects. Although definitive diagnosis of Lynch syndrome requires identification of a germline mutation in one of the MMR genes, this information from IHC testing can direct the germline testing to a specific MMR gene.
Table 24.2
Interpretation of protein loss patterns in MMR immunohistochemistry
Protein(s) lost | Possible gene defect a |
|---|---|
MLH1 and PMS2 | The most common pattern of loss. Both sporadic and hereditary tumors may show this pattern, but most are sporadic. MLH1 hypermethylation in sporadic tumors. MLH1 mutation in hereditary tumors. |
MSH2 and MSH6 | The most common exclusively hereditary pattern. MSH2 mutation. |
MSH6 only | Uncommon. MSH6 mutation. |
PMS2 only | Rare. PMS2 mutation or, less likely, MLH1 mutation. |
Although IHC should ideally clearly show that a MMR protein is either present or absent in a tumor, IHC results can be ambiguous. Tumor cells often show variable staining, making interpretation difficult, and even tumors that harbor MMR gene mutations may still demonstrate detectable protein staining [78–80]. One MLH1 truncation mutation has been reported to avoid degradation and stains positively by IHC, while its partner PMS2 is destabilized and shows protein loss, a pattern which would normally indicate PMS2 mutation [81]. It has been suggested that IHC staining for only two MMR proteins, MSH6 and PMS2, may be as effective as staining for all four MMR proteins as a screening test, since one of these should be lost in any of the most common Lynch syndrome scenarios [82]. This has a cost advantage, but many laboratories may choose to use all four stains to help clarify difficult cases and to provide immediate guidance for gene testing.
Sometimes, a tumor that is otherwise completely negative for a protein may show scattered positive nuclei. These may actually represent staining of benign lymphocytes admixed with tumor, the intratumoral lymphocytes (ITL) that are one of the histologic features of MMR-deficient tumors. One feature that can distinguish this artifact from true tumor staining is if the staining is limited to uniform, small, round nuclei characteristic of lymphocytes. Occasionally, apoptotic cells may aberrantly stain for a MMR protein that is absent in the tumor. It is often helpful to review the histologic appearance of the tumor for the presence of ITL and apoptotic cells. Another potential source of misinterpretation is diminished MSH6 staining in rectal carcinomas that have been treated with chemoradiation. Although the staining may be completely absent in these tumors, some may not have an MSH6 mutation or evidence of MSI. If a pretreatment specimen is available, staining is likely to be more representative of the true MSH6 status [83]. Finally, the interpretation of IHC testing should be reported as having MMR proteins “present” or “absent,” rather than positive or negative, to avoid possible misinterpretation by the recipient of the test results; “positive” may be incorrectly read as “positive for MMR deficiency.”
Microsatellite Instability PCR
PCR testing of DNA microsatellites represents an indirect functional assay for MMR proteins. After DNA is extracted from tumor tissue and from a non-tumor control tissue from the same patient, a panel of microsatellites is amplified by PCR using primers adjacent to but not including the repetitive sequence. The size of the PCR products is then analyzed by capillary electrophoresis, or less commonly by gel electrophoresis. Because defective MMR causes frameshift mutations in microsatellites, instability is shown by a change in the size of the PCR products from the microsatellite. Commonly used panels of microsatellites for MSI testing in the USA are the NCI panel (three dinucleotide [D2S123, D5S346, and D17S250] and two mononucleotide [Big Adenine Tract, BAT–25and BAT-26] microsatellites) and the panel supplied in a commercial kit available from Promega (Madison, WI) with five mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24, and MONO-27) [84]. Different microsatellites vary in their ability to identify tumors with defective MMR, with mononucleotide repeat loci generally superior to dinucleotide loci in both sensitivity and specificity [85]. This may be particularly true with MSH6 deficiency, which preferentially causes errors in mononucleotide repeats, as described earlier. The Promega panel includes two additional pentanucleotide repeats which are not evaluated to identify MSI but are used to ensure correct pairing of a patient’s tumor and normal DNA. These repeats are highly polymorphic in the population, and different patterns seen between a patient’s samples raise the possibility of specimen contamination or a labeling error.
As established by criteria from the National Cancer Institute, tumors exhibiting stability in all of the microsatellites tested are called microsatellite stable (MSS). Those with instability in ≥30–40 % of tested microsatellites are designated microsatellite instability-high (MSI-H). The third category, microsatellite instability-low (MSI-L), encompasses tumors that have at least one unstable microsatellite, but not enough to meet MSI-H criteria [86]. In common practice with the 5-microsatellite Promega or NCI panels, MSS, MSI-L, and MSI-H correspond to zero, one, or two or more unstable microsatellites, respectively. MSS tumors usually have intact MMR function, while MSI-H tumors correlate with MMR deficiency. MSI-L tumors have not been shown to be associated with defective MMR and are usually regarded as equivalent to MSS in the absence of solid evidence for Lynch syndrome. Instability is defined by a change in the length of a microsatellite in tumor DNA when compared to non-tumor (“normal”) DNA from the same patient. When compared with the normal control, an unstable allele will appear as a novel allele or a shift in the length of the allele.
The types of patterns seen in MSI testing depend upon whether dinucleotide or mononucleotide repeats are being evaluated. Dinucleotide repeats are polymorphic in the population, and a patient will often have alleles of different sizes on the maternal and paternal chromosomes. Thus, the normal sample will show two peaks, each representing one allele. One or both of these may change in length in the tumor as a result of defective MMR. The presence of two peaks in the normal DNA may mask a shift if the novel allele is obscured by an overlapping normal allele. Mononucleotide repeats, on the other hand, are generally very homogeneous (“quasi-monomorphic”), with same-sized alleles inherited from each parent. Thus, the normal sample will typically show only a single peak for each microsatellite. Tumor instability will be demonstrated as a shift in length for that peak or the presence of a second peak that was not seen in the normal tissue. Examples of unstable mononucleotide microsatellite alleles are shown in Fig. 24.3.
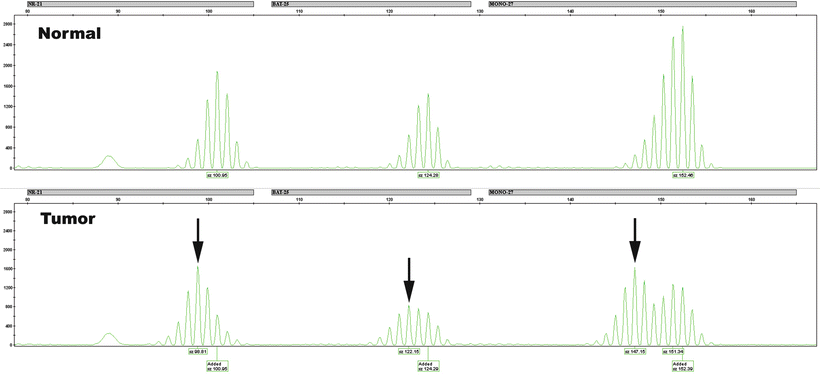
Figure 24.3
Three microsatellites with instability. Capillary electrophoresis data for three microsatellites, NR-21, BAT-26, and MONO-27, is shown for a patient’s normal (above) and tumor (below) DNA. Arrows point to changes in the size of microsatellites in the tumor, indicative of stability. Note that the subtle 2-bp size change in NR-21 and BAT-26 may be difficult to appreciate unless a comparison with the normal sample is made
The effectiveness of MSI testing is closely related to the panel of microsatellites used in the assay. Poor performance of some markers may result in MSI-L designation, and when many microsatellites are tested, the percentage of MSI-L tumors increases [87]. The percentage of MSI-L tumors in older reports, before the use of only mononucleotide panels, is as high as 35 % [88]. In contrast, when mononucleotide repeats are used exclusively, MSI-L tumors become very uncommon [89]. The significance of MSI-L when testing with a mononucleotide panel is unclear, and these patients should be offered genetic counseling. Fortunately, the percentage of unstable microsatellites in MSI-H tumors is usually 80–100 % when mononucleotide repeats are used [90]. Note that the pentanucleotide repeats in the Promega panel may show instability themselves in MSI-H tumors, and this can cause confusion if used to match a patient’s tumor and normal samples [89].
Recognition of some general patterns of instability in mononucleotide microsatellites can be helpful in MSI interpretation. First, mononucleotide markers nearly always shift to a smaller size when unstable. While larger sizes can be seen, their presence may indicate another genetic event such as an insertion, rather than instability. Second, the magnitude of a shift is usually no greater than approximately 10–12 bases, and large shifts often have some intermediate-sized alleles present as well. These intervening peaks are due to heterogeneous instability in the tumor. Shifts greater than approximately 12 bases may be the result of a deletion, and a deletion would not be expected to show the intermediate-sized alleles. A third pattern to be aware of is heterozygosity for a mononucleotide microsatellite. While most individuals have inherited alleles of the same size for a mononucleotide microsatellite, occasionally, patients have two different-sized alleles. This will be evident in both tumor and normal samples. Because heterozygosity is uncommon (only occurring in up to 5 % of cases), it is unlikely to be present in more than one or two microsatellites, and cases with more than this should be carefully evaluated for the possibility of specimen mislabeling or contamination of the normal DNA sample with tumor DNA. Finally, the minimum size of a shift is one base, the length of the repeating unit. Since identification of such a small shift is difficult, some clinical laboratories require a minimum of a two-base shift to score a microsatellite unstable. Fortunately, if a mononucleotide panel is being used, most MSI-H tumors will show 3–5 clearly unstable microsatellites.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


