Type
OMIM#
Gene
Chr.
Protein
Inheritance
Age of onset
1
235200
HFE
6p22.2
Hereditary Hemochromatosis Protein
Autosomal recessive
Adult
2A
602390
HJV
1q21.1
Hemojuvelin
Autosomal recessive
Child to young adult
2B
602390
HAMP
19q13.12
Hepcidin
Autosomal recessive
Child to young adult
3
604250
TFR2
7q22.1
Transferrin Receptor Protein 2
Autosomal recessive
Young adult to adult
4A
606069
SLC40A1
2q32.2
Solute carrier family 40 member 1 (Ferroportin)
Autosomal dominant (LOF)
Adult
4B
606069
SLC40A1
2q32.2
Autosomal dominant (GOF)
Adult
The HFE protein, a 343 amino acid transmembrane protein, is homologous to major histocompatibility class I molecules and associates with β2-microglobulin. The HFE protein is found predominantly in hepatocytes. At the cell surface, HFE complexes with the transferrin receptor protein 1 (TfR1) and competes for binding of iron-loaded transferrin. Under conditions of high iron, HFE is displaced from TfR1 and binds to transferrin receptor protein 2 (TfR2). Although the mechanism is not understood, the HFE-TfR2 complex is believed to create an intercellular signal to increase hepcidin expression. The role of HFE in HHC and iron homeostasis is supported by the fact that an HFE knockout mouse develops iron overload similar to that seen in human HHC [5].
Two allelic variants of the HFE gene, p.C282Y (c.845G>A) and p.H63D (c.187C>G), are significantly correlated with HHC, and most of the clinical studies have focused on these variants [6]. These two variants are usually not found on the same chromosome. The p.C282Y mutation abolishes a conserved disulfide bond which disrupts secondary structure required for HFE-binding to β2-microglobulin and subsequent transport to the cell surface. The p.C282Y HFE protein remains in the Golgi rather than being transported to the cell surface. The p.H63D protein product is expressed on the cell surface, but likely has an altered interaction with the transferrin receptor. Several other HFE allelic variants have been described, but little is known regarding their phenotypic effects. The p.S65C mutation has been implicated in HHC but its clinical significance is still unclear [2].
The frequency of the p.C282Y and p.H63D mutations in different ethnic groups has been reviewed [2]. These mutations are most common in populations of European descent. A pooled analysis of several Caucasian population studies found that the frequency of p.C282Y homozygosity was 0.4 % and heterozygosity was 9.2 % (Table 16.2). The frequency of p.H63D homozygosity was 2 % and heterozygosity was 23 %. Compound heterozygosity (p.C282Y/p.H63D) was 2 %. The frequency of HFE genotypes in HHC cases was 77.5 % for p.C282Y/p.C282Y, 5.3 % for p.C282Y/p.H63D, and 1.5 % for p.H63D/p.H63D. The pooled odds ratio (OR) for each HFE genotype indicated that individuals with the p.C282Y/p.C282Y and p.C282Y/p.H63D genotypes have the highest risk for iron overload with an OR of 4,383 and 32, respectively [7]. Individuals with the p.H63D/p.H63D and p.C282Y heterozygote (p.C282Y/+) genotypes are not considered to be at elevated risk for developing iron overload. The high frequency of HHC patients with the p.C282Y/p.C282Y genotype suggested that the penetrance for this genotype was very high. However, additional studies designed to estimate the penetrance suggested that the majority of p.C282Y homozygotes would not develop clinical disease resulting from iron overload. In a large prospective study, the proportion of p.C282Y homozygotes with documented iron-overload was 28.4 % for men and 1.2 % for women [8]. Currently, the estimates of the penetrance range from 1 % to 50 %. The penetrance of the p.C282Y/p.H63D genotype is much lower, ranging from 0.3 % to 1.4 %.
Clinical Utility of Testing
Diagnosis of HHC is based on clinical, biochemical, histological, and molecular studies, specifically tests such as transferrin saturation, ferritin concentration, evaluation of iron stores by liver biopsy, and genotyping for p.C282Y and p.H63D mutations in the HFE gene. Due to the reduced penetrance of p.C282Y/p.C282Y, the even lower penetrance of p.C282Y/p.H63D, and the presence of other mutations within HFE or in other genes related to HHC, the diagnosis or rule out of HHC cannot be made with a molecular test alone. However, in patients with clinical and biochemical symptoms of iron overload, molecular testing can be useful in confirming the diagnosis of HHC and may eliminate the need for liver biopsy. Typically, molecular testing is warranted in patients with transferrin saturation of >45 %. A genotyping result of p.C282Y/p.C282Y or p.C282Y/p.H63D is considered to confirm a diagnosis of HHC. A small number of cases (<5 %) will be identified as p.C282Y heterozygotes. It is possible these patients have other mutations in HFE or mutations in other genes involved in iron overload (Table 16.1).
Molecular testing also is used for carrier analysis and to identify relatives at risk for HHC. After the diagnosis of HHC is made in the proband, testing may be appropriate for other family members who may be at higher risk than the general population for developing iron overload. The iron status of these individuals can be monitored as appropriate. The HFE genotyping test can be used to identify individuals with the same genotype as the proband; these individuals would be at higher risk for developing iron overload than family members who did not inherit the mutations present in the proband. Requests for prenatal testing for HHC are highly unusual since HHC is a treatable adult onset disorder with low penetrance.
Soon after the identifications of the role of HFE mutations in iron overload, population screening for p.C282Y and p.H63D was considered. Since simple phlebotomy can prevent iron overload and its severe clinical complications, early identification of individuals at risk would be beneficial. However, given the low penetrance of the HFE genotypes, the majority of individuals identified with HFE mutations would be unlikely to develop clinical symptoms associated with HHC and yet might be subject to stigmatization and/or genetic discrimination as a result of the testing. At this time, population screening for HHC is not recommended due to these limitations.
Available Assays
As the p.C282Y and p.H63D mutations are single nucleotide polymorphisms (SNPs), they can be accurately detected using a variety of methods. As with all SNP assays, careful consideration must be given to the known sequence variants that may affect the assay method. The first method described for these mutations was an oligonucleotide ligation assay [6]. The primers described by this group to amplify the regions of the HFE gene have been used in a variety of assays. However, polymorphisms in one primer binding site sequence has been described and has the potential to result in a diagnostic error [9, 10]. Using primers that do not include the polymorphisms eliminates this potential error. The possible interference of the p.S65C mutation with the detection of p.H63D also should be considered in assay design. A number of additional assay methods for these mutations have been described, including PCR-RFLP, AS-PCR, ASO hybridization, single-nucleotide primer extension, and real-time PCR methods [11].
A simple method for detection of p.C282Y and p.H63D is PCR-RFLP. p.C282Y is detected by digestion of the PCR product with RsaI, and H63D is detected by using BclI or MboI. The digested PCR products are analyzed by gel electrophoresis. The advantage of this method is that optimization is relatively straightforward and expensive equipment is not required. The disadvantage is a longer turnaround time for testing. AS-PCR assays distinguish the normal and mutant alleles directly with only gel electrophoresis of the PCR products, so the post-PCR processing is reduced. The AS-PCR reactions require careful optimization, and multiple reactions are required to determine the genotype.
Real-time PCR methods that couple PCR with detection of hybridization probes are commonly used to detect single nucleotide polymorphisms. These techniques are rapid, sensitive, have a wide dynamic range, and require no additional processing after PCR. A variety of chemistries are available such as FRET hybridization probes with melt curve analysis, hydrolysis probes, and molecular beacons. For each method, the appropriate negative and positive controls are required to demonstrate the specificity of the assay. A melting curve of the hybridization probe fluorescence is used to detect changes in thermal stability and therefore discriminate single base mutations in a single reaction (Fig. 16.1). An additional advantage of real-time PCR is that new sequence variations in the probe regions are detected. For example, the probe for p.H63D also detects the p.S65C mutation [12]. In cases where testing is indicated for additional HFE mutations or HHC types 2–4, HFE, HJV, HAMP, TFR2, and SLC40A1 are analyzed by DNA sequencing.
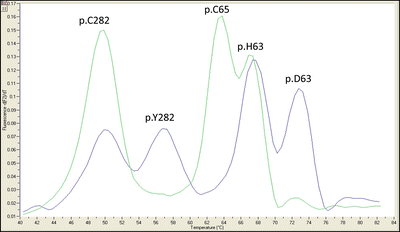
Figure 16.1
PCR-FRET hybridization probe with melt curve analysis for p.Cys282Tyr (p.C282Y) and p.His63Asp (p.H63D) alleles of the HFE gene. One set of probes distinguishes the p.C282Y alleles and the other set p.H63D alleles. The probes for p.H63D also overlap the p.S65C allele so it can also be identified. Each allele has a specific melting temperature. Since the melting temperatures of the probes do not overlap, this assay can be performed using a single fluorophore. An example of a p.C282Y/p.H63D compound heterozygous sample (blue trace) and a sample negative for p.C282Y and p.H63D and positive for p.S65C (green trace) are shown
Interpretation of Test Results
The analytic sensitivity for detecting these mutations should be >99 %. False positives and false negatives are rare. However, sequence variations in primers, probes, and restriction sites have the potential to lead to a diagnostic error. A polymorphism reported at the 3′ end of one of the primers described in the original publication [6] was later reported to prevent the amplification of wild-type p.C282Y alleles with some assay methods [9].
Interpretations should be made in the context of all clinical information and account for the reduced penetrance and variable expression observed with HHC. In individuals with biochemical evidence of iron overload, a genotype of p.C282Y/p.C282Y or p.C282Y/p.H63D confirms a diagnosis of HFE-related HHC. Up to 5 % of these individuals will be heterozygous for p.C282Y. Testing for other HFE mutations or mutations in other iron-related genes may be indicated. Individuals who test negative for p.C282Y are a heterogeneous group with other genetic causes of HHC or iron overload caused by liver disease or a metabolic disorder. Unaffected individuals with the p.C282Y/p.C282Y genotype have an increased risk of HHC with a penetrance of 1–50 %. p.C282Y heterozygotes may have elevated iron levels but rarely develop complications of iron overload.
Genetic counseling should be recommended when p.C282Y and p.H63D are identified in a family. Molecular testing will be useful in identifying carriers, homozygous individuals, and those who did not inherit the at-risk genotype. Most parents of the proband will be p.C282Y heterozygotes so the risk to siblings of a proband is 25 % but could be as high as 50 % if one of the parents is homozygous. The risk assessment should include the most current estimates of the disease penetrance.
Laboratory Issues
Proficiency testing for p.C282Y and p.H63D mutations in the HFE gene is available through the College of American Pathologists (CAP) Molecular Genetics Laboratory (MGL) survey. Cell-line controls can be purchased from Coriell Cell Repositories (http:/ccr.coriell.org/).
Hemoglobinopathies
Molecular Basis of Disease
Hemoglobinopathies are the most common single-gene diseases in the world; approximately 5–7 % of the world’s population is a carrier for one of the hemoglobin disorders. The extensive study of these disorders has led to much of our knowledge regarding protein structure and function, mutational mechanisms, gene expression during development, and evolution. Extensive reviews of these disorders are available [13, 14]. A list of the numerous characterized mutant alleles is available at the Globin Gene Server (http://globin.cse.psu.edu) and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
Hemoglobin (Hb) functions as an oxygen carrier in vertebrate red cells. The protein is a tetramer consisting of two α and two β chains. Each subunit contains a heme prosthetic group that is responsible for the oxygen-binding capacity of the protein. These chains are highly homologous to one another at the primary sequence level as well as in three-dimensional structure. Six different globin chains (α, β, γ, δ, ε, ζ) are found in normal human hemoglobins at various times during development [13–15]. The α and β chains are encoded by separate genetic loci (Fig. 16.2). The α and α-like chains (ζ, Ψζ, Ψα) are clustered on chromosome 16. In addition, two copies of the α-globin genes, α1 and α2, are located on each chromosome 16. The β and β-like chains (ε, Gγ, Aγ, Ψβ, δ) are clustered on chromosome 11.
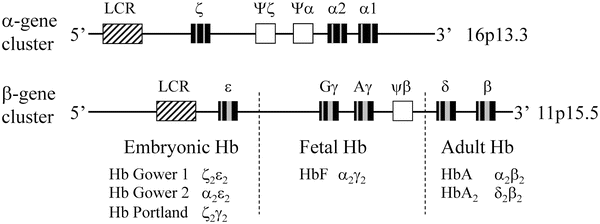
Figure 16.2
Genomic organization and developmental expression of the α- and β-globin gene clusters. The boxes identify the locus control region (LCR; diagonal), pseudogenes (white), exons (black), and introns (gray). The developmental time periods for the expression of the β-globin genes are indicated by the dashed lines. (Adapted from Stamatoyannopoulos G, Nienhuis AW. Hemoglobin switching. In: Stamatoyannopoulos G, Nienhuis AW, Majerus PW, Varmus H, eds. The molecular basis of blood diseases. Philadelphia: W.B. Saunders, 1994:107–55)
The developmental regulation of these genes is important to the understanding of many of the hemoglobin disorders. The gene order in each cluster is identical to the order of expression during development. In each developmental stage, there is an equimolar production of α and β chains. Hb Gower 1 (ζ2ε2), Hb Gower 2 (α2ε2), and Hb Portland (ζ2γ2) are expressed in embryos. In the fetal stage, the Hb is predominantly α2γ2 (HbF). In normal adults, the Hb is comprised of approximately 96 % α2β2 (HbA), 2.5 % δ2β2 (HbA2), and approximately 1 % α2γ2 (HbF). In addition to promoters and enhancer elements, the locus control region (LCR) is responsible for developmental expression of these genes. The LCR for the β locus is approximately 20 kb upstream of the ε-globin gene and the LCR for the α locus is approximately 40 kb upstream from the ζ-globin gene. The LCR is required for expression of all the globin genes at the locus [13].
The difference in gene dosage and expression patterns between α and β genes is significant. Each diploid genome encodes four copies of the α gene and two copies of the β gene. Since γ globin is expressed during fetal life and shortly after birth, β globin mutations do not exert their deleterious effects during the prenatal period. In contrast, since α chains are expressed during fetal and postnatal life, mutations in the α chain can cause severe disease during both developmental stages.
The hemoglobinopathies are commonly divided into two broad categories: structural variants, and thalassemias that are characterized by reduced rates of production of α or β chains [13]. These categories are not exclusive, as structural variants, which result in unstable hemoglobins, can be associated with both α- and β-thalassemia phenotypes. Although an extensive number of abnormal hemoglobins have been described, this chapter focuses on two of the most common structural variants, sickle hemoglobin (HbS) and HbC, and the more common forms of α- and β-thalassemia.
Sickle hemoglobin results from a single nucleotide substitution (GAG→GTG) that changes codon 6 of the β-globin gene from a glutamic acid to a valine (p.Glu6Val). Using standard nomenclature this variant should be reported as p.Glu7Val; however, the extensive literature uses the numbering of the mature protein in which the first methionine is cleaved. Homozygosity for this mutation causes SD, an autosomal recessive disorder. SD is a major cause of morbidity and mortality in Africa and in populations with individuals of African descent such as the Mediterranean area, the Middle East, and India [13]. Under the condition of low oxygen tension in the microvasculature, HbS polymerizes into fibers that cause the erythrocytes to sickle. The poor deformability of the sickle red cells and their defective passage in the micro-circulation causes the vasoocclusive events that are the hallmark of SD. The shortened survival of these red cells leads to a chronic hemolytic anemia. The frequency of SD is 1 in 600 in African Americans; the heterozygote frequency in African Americans is approximately 8 %, but the frequency in Africa can be much higher [13]. The frequency of the HbS allele, as well as alleles causing thalassemia, is maintained by their protective effect against malaria. The heterozygous state, sickle cell trait, is clinically normal but may be at risk for vasoocclusive events when exposed to low oxygen tension such as flying at high altitude in an airplane with reduced cabin pressure.
The sickling disorders also include compound heterozygous states with the HbS mutation in association with another β-globin variant such as HbC, or β-thalassemia. Similar to HbS, HbC also has an amino acid substitution at position 6 of the β-globin gene (Glu6Lys). HbC is less soluble than HbA and leads to reduced deformability of the red cells and a mild hemolytic anemia. The allele is frequent in persons of African descent; approximately 3 % of African Americans are carriers [13]. Individuals with βSβC have sickle cell hemoglobin (SC) disease. SC disease is a milder hemolytic disorder than SD and therefore may go unrecognized until a serious complication occurs.
The thalassemias result from the reduced synthesis or stability of either the α- or β-globin chain. The chains in excess due to the α:β imbalance precipitate within the red blood cells, leading to membrane damage and red cell destruction. The defect produces a hypochromic, microcytic anemia. The population distribution of thalassemia includes the Mediterranean, Middle East, portions of Africa, India, and Southeast Asia; however, the migration of these populations has resulted in the worldwide occurrence of these diseases [13].
The most common forms of α-thalassemia are caused by deletions. The genetics of α-thalassemia are complicated by the fact that each normal chromosome has two α genes and that there are haplotypes with either the loss of one (-α) (α+ thalassemia) or both (–) (αo thalassemia) copies of the α gene. In addition to the normal genotype, four additional genotypes are possible: silent carrier (αα/-α), α-thalassemia trait (-α/-α or –/αα), HbH (β4) disease (-α/–), and hydrops fetalis, Hb Barts: γ4 (–/–). Individuals with HbH disease have a moderately severe hemolytic anemia, while the inheritance of no copies of the α gene (Hb Barts) is incompatible with life. Carriers of α-thalassemia trait have only a mild hypochromic microcytic anemia, but couples with these genotypes are at risk for having a fetus with hydrops fetalis or a child with HbH disease. The (–) genotype, and therefore the risk of hydrops fetalis, is largely restricted to Southeast Asia, while in other groups the (-α) genotype is more common [13, 14]. Distinguishing these two carrier states is important for accurate genetic counseling. The most widely occurring single α-globin gene deletions are -α3.7 and -α4.2. The double α-globin gene deletions α-SEA, α-FIL, and α-THAI are common in Southeast Asia while α-MED and -(α)20 are more common in the Mediterranean area. Nondeletion α-thalassemia alleles are rare, but the αConstant Spring, a mutation in the termination codon of the α2 gene, is frequently present in Southeast Asia.
The β-thalassemias are mainly inherited in an autosomal recessive manner, although there are some dominant forms. Heterozygotes are asymptomatic but with recognizable hematologic parameters including an elevated HbA2. Homozygotes and compound heterozygotes of HbE develop a severe anemia within the first year of life as the switch from γ to β chains occurs. In contrast to α-thalassemia, β-thalassemias are heterogeneous at the molecular level. The molecular mechanism of the mutations is equally varied. Most mutations are single-nucleotide substitutions or frameshift mutations. More than 200 disease-causing mutations have been identified in the β-globin gene; however, within each at-risk population there is a set of 4–10 mutations that account for the majority of disease-causing alleles (Table 16.3) [15]. HbE is a β-globin variant (Glu26Lys) that can cause a mild thalassemia phenotype. It is one of the most common Hb variants, and the frequency is high in Southeast Asia. Although HbE homozygotes are asymptomatic, compound heterozygotes with another β-thalassemia allele will have an abnormal phenotype [13]. Although deletions are rare, a 619 base pair (bp) deletion involving the 3′ end of the β-globin gene is common in India and Pakistan, accounting for approximately 30 % of the β-thalassemia alleles [13]. β-Thalassemia also may result from deletion of part of the globin gene cluster (e.g., 5P-thalassemia) or from deletions that start 50–100 kilobases (kb) upstream from the globin gene cluster and extend 3′ into the cluster (εγδβ-thalassemia). Some of these deletions lead to hereditary persistence of fetal hemoglobin. In addition, there are fusion chain variants such as Hb Lepore, a δβ+ thalassemia. Mutations either completely (β°-thalassemia) or partially (β+-thalassemia) abolish β-chain production. Milder phenotypes have been recognized that can be explained at the molecular level by homozygosity or compound heterozygosity for mild or silent mutations, coinheritance of α-thalassemia, or coinheritance of a genetic determinant that increases the production of the γ chain.
Table 16.2
HFE genotype frequencies and their contribution to hereditary hemochromatosis
Genotype | Population frequency (%) | Pooled odds ratio | 95 % CI | % HHC cases |
|---|---|---|---|---|
p.C282Y/p.C282Y | 0.4 | 4,383 | 1,374–>10,000 | 77.5 |
p.C282Y/p.H63D | 2.0 | 32 | 18.5–55.4 | 5.3 |
p.H63D/p.H63D | 2.0 | 5.7 | 3.2–10.1 | 1.5 |
p.C282Y/+ | 9.2 | 4.1 | 2.9–5.8
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|