Marker
Indication
1p/19q deletion
Oligodendroglioma diagnosis and prognosis
Prediction of oligodendroglioma therapeutic response
Confirmation of an oligodendroglial component in oligoastrocytomas
MGMT promoter methylation
Response of glioblastoma to alkylating drugs (temozolomide)
Small cell glioblastoma diagnosis
Prognostic marker
IDH1 mutation
Diagnostic tool for low- and intermediate-grade astrocytomas and oligodendrogliomas
Distinction from primary glioblastomas, pilocytic astrocytomas, and ependymomas
Neoplastic glioma vs reactive gliosis
Prognostic marker
EGFR amplification (overexpression)
Differentiation from oligodendroglioma
Therapeutic response to novel EGFR pathway inhibitors
Chromosome 10 loss/PTEN mutation
Prognosis of anaplastic astrocytomas
Response to EGFR pathway inhibitors
TP53 mutation
Secondary glioblastoma vs primary
Neoplastic glioma vs reactive gliosis
Pathology Considerations
Gliomas are classified into tumors of astrocytic, oligodendroglial, ependymal, and mixed lineage. “Diffuse gliomas” is a descriptive general term that applies to all diffusely infiltrating tumors of glial lineages before a more specific determination of lineage can be determined based on molecular and immunohistochemical markers. Diffuse astrocytic tumors are divided into three different grades by the World Health Organization (WHO) system [3]: grade II (or astrocytoma [A]), grade III (or anaplastic astrocytoma [AA]), and grade IV (or glioblastoma, previously known as glioblastoma multiforme [GBM]). These tumors occur predominantly in adults. Diffuse astrocytic tumors can be subclassified into several histological types, including fibrillary, gemistocytic, small cell, and giant cell subtypes. Several of these histological types can be found within a single diffuse astrocytic tumor. Pilocytic astrocytomas are nondiffuse, well circumscribed, and localized. Pilocytic astrocytomas are often WHO grade I, and predominantly occur in children and young adults. Glioblastomas can be subclassified into two groups based on the duration of symptoms [4]. Primary glioblastoma presents de novo with a short duration of symptoms (often less than 3 months). Secondary glioblastoma presents with a longer duration of symptoms or with a preceding grade II or III glioma. Primary and secondary glioblastomas usually present in patients greater than 60 and less than 40 years of age, respectively.
Oligodendrogliomas most commonly are divided into two grades: grade II (oligodendroglioma) and grade III (anaplastic oligodendroglioma), although some neuropathologists have described a grade IV oligodendroglioma [5]. Ependymomas are also classified as low (WHO grade II) and high grade (anaplastic or WHO grade III).
One of the problems with the neuropathologic diagnosis of gliomas is their histologic heterogeneity. AA exhibits increased cellularity, nuclear atypia, and mitotic activity. Microvascular proliferation and necrosis are additional features of glioblastomas. Depending on the extent of surgical sampling, some of the histologic elements that distinguish AA and glioblastoma may not be present in the specimen submitted for pathological diagnosis, and some gliomas may be undergraded. Thus, molecular markers that assist in tumor grading are clinically useful.
In addition to histologic heterogeneity, diffuse gliomas often exhibit cell type heterogeneity. Both astrocytic and oligodendroglial elements may be present in diffuse gliomas. When both of these elements comprise a significant proportion of the tumor, the glioma used to be classified as a mixed oligoastrocytoma (MOA). However, this term no longer exists based on recent recommendations of the International Society of Neuropathology [6].
Molecular Pathology Tools for Glioma Analysis and Grading
Gliomas have been extensively characterized by cytogenetic, molecular cytogenetic, and molecular genetic methods (reviewed in Refs. 4, 7, 8). Figure 38.1 summarizes the major genetic alterations found in diffuse gliomas. Some of these alterations are highly correlated with the grade of gliomas, especially for astrocytic tumors.
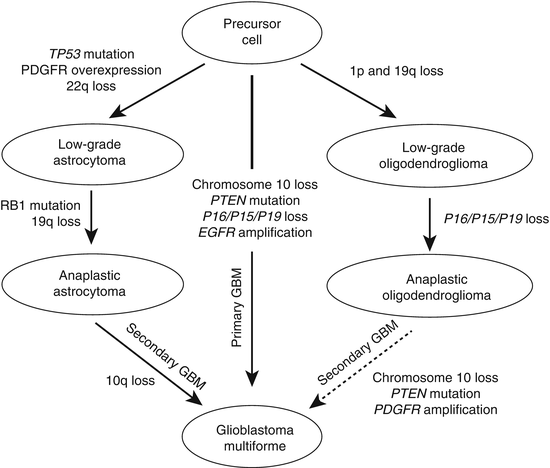
Figure 38.1
Possible genetic pathways of diffuse glioma formation and progression (modified from Refs. 7 and 8). Histologic classification is based on current WHO guidelines [3]. Although the molecular genetic evidence is increasing, it is still controversial whether anaplastic oligodendroglioma progresses to glioblastoma (GBm) (dashed arrow)
Several genetic alterations have been associated with gliomas, especially primary glioblastoma and oligodendrogliomas. These alterations include chromosomal deletions of chromosome arms 1p and 19q, MGMT promoter methylation, loss of chromosome 10, PTEN mutations, IDH1 mutations, and amplification of EGFR with resulting overexpression of the EGFR protein. While the clinical use of these markers has been validated only partially, growing evidence supports the usefulness of these molecular markers for glioma grading, prognosis, and treatment decisions. The presence of EGFR amplification, chromosome 10 loss, or PTEN mutation, or some combination of the three, in a glioma indicates that the tumor is likely a glioblastoma. Importantly, a small but significant proportion of tumors with the histologic features of AA have one or more of these alterations [9–13]. It is reasonable to hypothesize, though needs to be proven, that since patients with glioblastoma have a poor prognosis, then patients whose AA shares molecular characteristics with glioblastoma also may have poor survival.
One of the first and most significant molecular developments in the study of gliomas was the discovery of chromosomal alterations in chromosomes 1 and 19. Cairncross et al. demonstrated in 1994 that some anaplastic oligodendrogliomas respond to combination chemotherapy and radiation therapy [14]. Simultaneously, several investigators observed that approximately 70 % of oligodendrogliomas have deletions of 19q, 70 % have deletions of 1p, and 60 % have deletions of both 1p and 19q [7]. Usually, the entire chromosomal arms are deleted [15]. In contrast, about 40 %, 20 % and 10 % of astrocytomas have deletion of 19q, 1p, and both 1p and 19q, respectively [15]. The proportion of MOA with 1p and 19q deletions is intermediate, suggesting that some of these tumors can be grouped with oligodendrogliomas [15, 16]. A significant proportion of MOA also has TP53 mutations and other genetic anomalies, suggesting that these tumors can be grouped with the astrocytomas [15, 16].
Two retrospective studies have provided evidence that the oligodendrogliomas with 1p deletions, 19q deletions, or both have a better prognosis and respond better to chemotherapy and radiation therapy [17, 18]. The data strongly suggest that 1p/19q deletions are associated with tumors of oligodendroglial lineage, with oligodendrogliomas that have a better prognosis, and with oligodendrogliomas that respond to chemotherapy and radiation therapy. Additional prospective studies provided evidence that the presence or absence of 1p/19q deletions suggests different treatment recommendations for oligodendrogliomas that require adjuvant therapy, especially anaplastic oligodendrogliomas [19, 20]. Patients with the deletion may receive chemotherapy first and delay radiation treatment, while patients without the deletion may receive radiation as first-line treatment.
Other alterations occur in oligodendrogliomas that lack 1p and 19q deletions. CDKN2A–CDKN2B–ARF (P16, P15, P19), PTEN, and chromosome 10 are frequently deleted in anaplastic oligodendrogliomas. The expression of CDKN2A and CDKN2B often is inactivated by promoter methylation. While the presence of these alterations is associated with a poorer prognosis [17, 21, 22], their detection has not become a part of standard clinical neurooncology.
MGMT is a DNA repair enzyme that normally counteracts the cell death effect of alkylating chemotherapy agents such as temozolomide, which is widely used in the treatment of glioblastoma. MGMT activity is downregulated and not fully expressed in about half of patients with glioblastoma due to MGMT gene promoter methylation and decreased gene expression and enzyme production. Thus, the presence of MGMT promoter methylation determines that the patient will be more responsive to chemotherapy. MGMT promoter methylation also correlates with better prognosis in general, suggesting that it might indicate a prognostically favorable molecular phenotype of the disease. In a study by Hegi et al., 46 % of the patients with MGMT methylation were alive at 2 years, while only 23 % of the patients with unmethylated MGMT were alive at 2 years [23]. Temozolomide provides some beneficial effect even in patients with unmethylated MGMT promoter; thus, both groups receive the same treatment. Since the methylation pattern might change over time, repeated testing at each tumor resection is justified.
Several growth factors are upregulated and involved in the pathogenesis of gliomas, especially the most malignant forms of glioblastoma. EGFR signaling is upregulated in approximately 30 % of gliomas and 60 % of glioblastomas. An even higher percentage of “small cell glioblastoma,” which can be misdiagnosed and possibly confused for oligodendroglioma, has upregulated EGFR signaling. The presence of EGFR upregulation as opposed to 1p/19q deletion may help to distinguish between glioblastoma and malignant oligodendroglioma, respectively. Another scenario in which EGFR overexpression is diagnostically useful is when a biopsy is taken from the edge of a tumor that appears to be histologically consistent with a grade III glioma but radiographically appears as glioblastoma. The presence of EGFR overexpression in these tumor specimens may support a diagnosis of glioblastoma instead of AA.
In glioblastomas that overexpress EGFR, mutant forms of EGFR are present in about half of the cases. The most common variant is EGFR vIII, which has deletion of exons 2–7 of the EGFR gene and results in an EGFR protein that is active independently of ligand binding (reviewed in Ref. 24). Several other amplified and mutated EGFR proteins also have been identified (e.g., the C958 variant is truncated after amino acid 958), some of which remain ligand dependent. Current translational studies are investigating the prevalence of these different amplified and mutated EGFR variants in primary and secondary glioblastoma and in glioblastoma from patients of different ages [25].
Many glioblastomas have dysregulation of EGFR downstream signaling, such as PTEN tumor suppressor gene mutations, phosphoinositide 3-kinase pathway, and AKT. These findings suggested a rationale for drug treatments that target these pathways. Mellinghoff and colleagues showed that gliomas with an EGFR vIII mutation in the presence of an intact PTEN-AKT pathway had increased sensitivity to erlotinib, whereas tumors with loss of the PTEN-AKT signaling did not. However subsequent trials showed less response to EGFR-targeted therapies, so the use of these drugs remains controversial.
Mutations in the IDH1 and IDH2 genes occur in high frequency in primary low- and intermediate-grade diffuse gliomas (both in astrocytomas and in oligodendrogliomas) [7, 20, 26, 27], as well as secondary glioblastomas. The most common mutation is the R132H mutation of IDH1, with a frequency of over 80 % in grade II astrocytomas, oligodendrogliomas, anaplastic oligodendrogliomas, and mixed oligoastrocytomas, close to 70 % of AA, but only less than 0.5 % in primary glioblastoma. IDH1 mutations also serve as a tool to distinguish astrocytomas and oligodendrogliomas from low-grade tumors that usually do not express this mutation such as ependymomas, pilocytic astrocytomas, and dysembryoplastic neuroepithelial tumors, as well as neoplastic glial proliferation from reactive gliosis. Gliomas with IDH1 mutations have a better prognosis, with or without adjuvant therapy. Therefore, IDH1 mutation serves as a prognostic and diagnostic marker (Fig. 38.2).
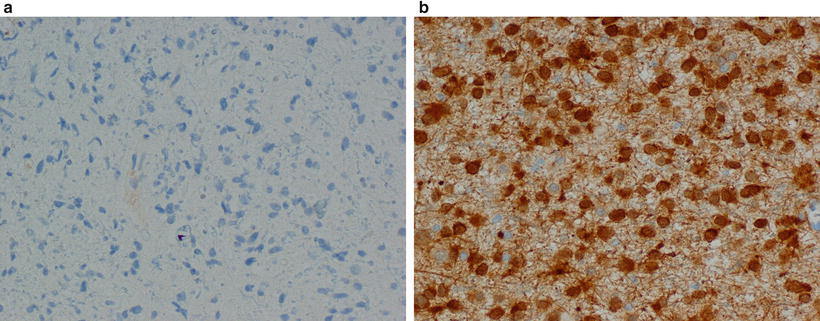
Figure 38.2
Immunohistochemistry images of low-grade glial neoplasms with negative (a) and positive (b) staining with antibody against the R132H mutations in IDH1
A Mayo Clinic/North Central Cancer Treatment Group (NCCTG) study indicated that AA with chromosome 10 loss or PTEN mutation, using full sequencing of the PTEN gene and exons 5–8 of the TP53 gene, behaves like glioblastomas [10]. The median survival of patients with AA and chromosome 10 loss or PTEN mutation was approximately 4 months, a survival worse than that of patients with glioblastoma (median survival of approximately 12 months). The median survival of patients whose AA lacked these alterations was 34 months. This difference in survival was statistically significant even after adjustment for patient age, performance score, and extent of resection, all of which are important prognostic variables for patients with gliomas. Importantly, the patient survival for glioblastoma with and without chromosome 10 loss or PTEN mutations was very similar [10]. It is possible that sampling bias (in terms of the tissue available for histologic and molecular analysis) may have accounted for the differences. However, sampling bias is a difficult problem to overcome in routine clinical practice, and molecular testing for chromosome 10 loss or PTEN mutations in a sample of a tumor with the features of AA indicates that the tumor may be a glioblastoma.
Interestingly, the Mayo Clinic/NCCTG study did not show that AA with EGFR amplification behaved like glioblastoma [10]. However, careful stratification indicated that AA and glioblastoma with EGFR amplification in young patients (i.e., < 40 years old) progress much faster than AA and glioblastoma without EGFR amplification in this age group [10]. Conversely, AA and glioblastoma with EGFR amplification in older patients (i.e., > 60 years old) had a better prognosis than those without this alteration. Importantly, this result has been confirmed by another group [28]. These differences in age-related prognosis may reflect the different biology of primary and secondary high-grade astrocytic tumors.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


