Chapter 4
Genes and Genetic Diseases

In the nineteenth century, microscopic studies of cells led scientists to suspect that the nucleus of the cell contained the important mechanisms of inheritance. Scientists found that chromatin, the substance that gives the nucleus a granular appearance, is observable in nondividing cells (also see Chapter 6). Just before the cell divides, the chromatin condenses to form microscopically observable, threadlike structures called chromosomes. (Cell division and chromosomes are discussed in Chapter 1.) With the rediscovery of Gregor Mendel’s important breeding experiments at the turn of the twentieth century, it soon became apparent that the chromosomes contained genes, the basic units of inheritance. Genes are composed of sequences of deoxyribonucleic acid (DNA), a primary constituent of chromosomes; the other primary constituent consists of proteins, such as histones, that cause the DNA to coil into a highly compressed structure (Figure 1-2, Chapter 1). By serving as the blueprints of proteins in the body, genes ultimately influence all aspects of body structure and function. Humans have approximately 20,000 to 25,000 genes. An error in one of these genes can lead to a recognizable genetic disease. The composition of genes at a given locus is known as the genotype. The outward appearance of an individual, which is the result of both genotype and environment, is the phenotype (p. 151).
To date, more than 21,000 human genetic traits have been identified and cataloged.1 About one third of pediatric inpatients are children with genetic diseases.2 In addition, many common diseases that affect primarily adults, such as hypertension, coronary heart disease, diabetes, and cancer, are now known to have important genetic components. (These diseases are also affected by environmental factors. The interaction between genetic and environmental components is discussed in Chapter 5.)
Great progress is being made in the diagnosis of genetic diseases and the understanding of genetic mechanisms underlying them. Genetic testing is used increasingly to guide drug choice and dosage, and gene therapy—the direct alteration of genes in cells—is now carried out effectively for some diseases. Genetics is now one of the most rapidly advancing fields of medicine (Boxes 4-1 and 4-2).
DNA, RNA, and Proteins: Heredity at the Molecular Level
DNA
Composition and Structure
Genes are composed of DNA and the most important constituent of DNA is four types of nitrogenous bases (Figure 4-1). The four bases, adenine, cytosine, guanine, and thymine, are commonly represented by their first letters: A, C, G, and T, respectively.
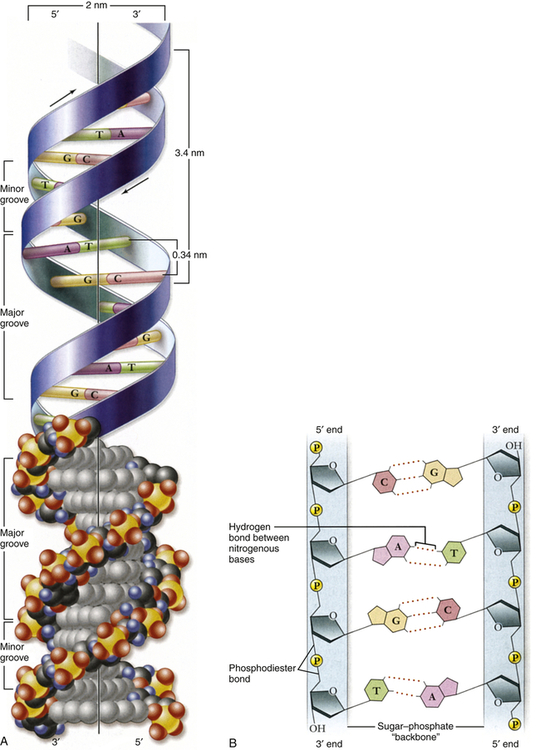
A, Double helix. Shown with the phosphodiester backbone as a ribbon on top and a space-filling model on the bottom. The bases protrude into the interior of the helix where they hold it together by base pairing. The backbone forms two grooves, the larger major groove and the smaller minor groove. B, Base pairing holds strands together. The H-bonds that form between A and T and between G and C are shown with dashed lines. These produce AT and GC base pairs that hold the two strands together. This always pairs a purine with a pyrimidine, keeping the diameter of the double helix constant. A, Adenine; C, cytosine; G, guanine; T, thymine. (From Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
In the early 1950s, James Watson and Francis Crick determined the physical structure of DNA. They proposed the now-famous double-helix model, in which DNA can be envisioned as a twisted ladder with chemical bonds as its rungs (see Figure 4-1). Projecting from each side of the ladder, at regular intervals, are the nitrogenous bases. The base projecting from one side is bound to the base projecting from the other by a weak hydrogen bond. Therefore, the nitrogenous bases form the rungs of the ladder; adenine pairs with thymine, and guanine pairs with cytosine. Each DNA subunit—consisting of one deoxyribose molecule, one phosphate group, and one base (see Figure 4-1)—is called a nucleotide.
DNA as the Genetic Code
Another significant feature of the genetic code is that it is nearly universal: with the exception of ciliated protozoa and some plants, all organisms use precisely the same DNA codes to specify proteins. An important general exception to this rule occurs in mitochondria—cytoplasmic organelles that are the sites of cellular respiration (see Chapter 1). The mitochondria have their own extranuclear DNA. Several codons of mitochondrial DNA encode different amino acids than do the same nuclear DNA codons.
Replication
In addition to having the ability to specify amino acid sequences, DNA must be able to replicate itself accurately during cell division if it is to serve as the basic genetic material. DNA replication consists of the breaking of the weak hydrogen bonds between the bases, leaving a single strand with each base unpaired. The consistent pairing of adenine with thymine and of guanine with cytosine, known as complementary base pairing, is the key to accurate replication. The principle of complementary base pairing dictates that the unpaired base will attract a free nucleotide only if the nucleotide has the proper complementary base. Thus a portion of a single strand with a sequence of bases labeled ATTGCT will bond with a series of free nucleotides with the bases TAACGA. When replication is complete, a new double-stranded molecule identical to the original is formed (Figure 4-2, A). The single strand is said to be a template (guide), or molecule on which a complementary molecule is built, and is the basis for synthesizing the new double strand.
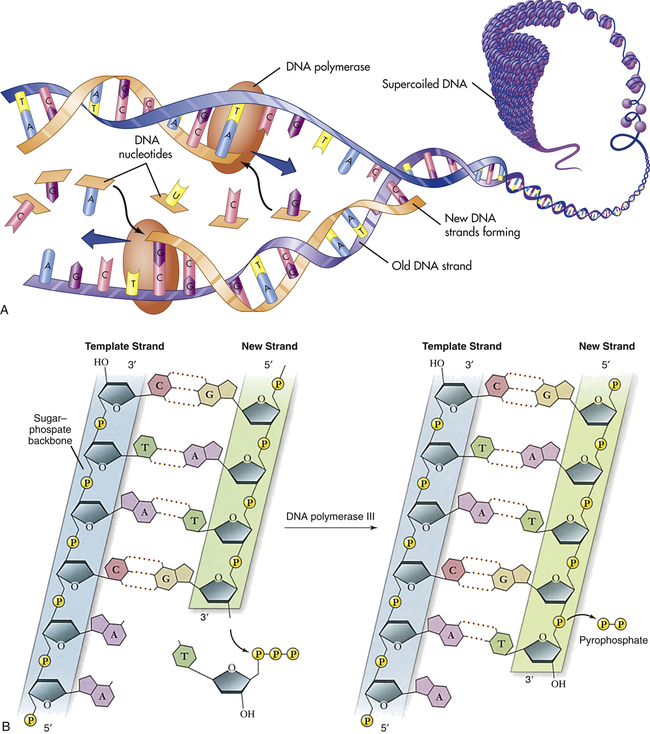
A, Replication of DNA. B, Action of DNA polymerase. DNA polymerases add nucleotides to the 3′ end of a growing chain. The nucleotide added depends on the base that is in the template strand. Each new base must be complementary to the base in the template strand. With the addition of each new nucleotide, triphosphate, two of its phosphates are cleaved off as pyrophosphate. A, Adenine; T, thymine; G, guanine; C, cytosine. (A from Patton KT, Thibodeau GA: Anatomy & physiology, ed 8, St Louis, 2013, Mosby; B adapted from Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Several different proteins are involved in DNA replication. One protein unwinds the double helix, one holds the strands apart, and others perform different distinct functions. The most important of these proteins is the enzyme known as DNA polymerase. This enzyme travels along the single DNA strand, adding the correct nucleotides to the free end of the new strand (see Figure 4-2, B). Besides adding the new nucleotides, the DNA polymerase performs a proofreading procedure. After the new nucleotide has been added to the chain, the DNA polymerase checks to make sure that its base is actually complementary to the template base. If it is not, the incorrect nucleotide is excised and replaced with a correct one. This procedure, one of the mechanisms of DNA repair, substantially enhances the accuracy of DNA replication.
Mutation
A mutation is any inherited alteration of genetic material. Microscopically observable alterations of chromosome number or structure are examples of mutations. Some mutations are much too small to be observed under a microscope. An example is the base pair substitution, in which one base pair is replaced by another. This mutation is sometimes called missense mutation because the “sense” of the codon produced after transcription of the mutant gene is altered (Figure 4-3). This substitution sometimes results in a change in amino acid sequence, but because of the redundancy of the genetic code, it may have no consequence. Profound consequences can result when an amino acid sequence is altered by a base pair substitution. (Many of the serious genetic diseases discussed later are the result of base pair substitutions.)
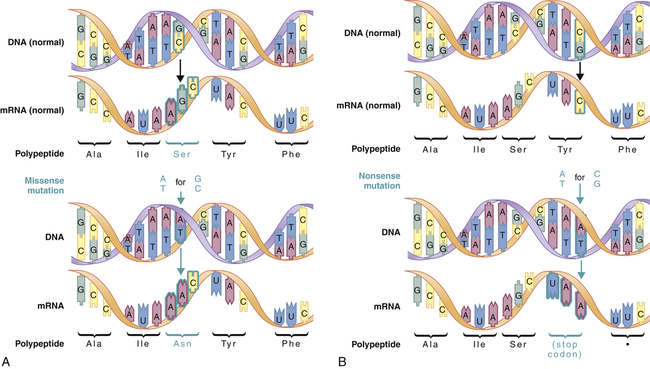
Missense mutations (A) produce a single amino acid change, whereas nonsense mutations (B) produce a stop codon in the mRNA. Stop codons terminate translation of the polypeptide. (From Jorde LB et al: Medical genetics, ed 4, St Louis, 2010, Mosby.)
A second major type of mutation is the frameshift mutation. This alteration involves the insertion or deletion of one or more base pairs to the DNA molecule. As Figure 4-4 shows, these mutations can change the entire “reading frame” of the DNA sequence because codons consist of groups of three base pairs. A frameshift mutation thus can greatly alter the resulting amino acid sequence.
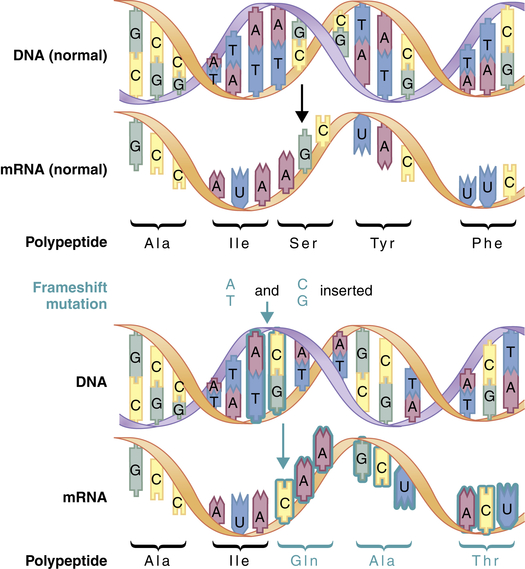
Frameshift mutations result from the addition or deletion of a number of bases that is not a multiple of 3. This mutation alters all of the codons downstream from the site of insertion or deletion. (From Jorde LB et al: Medical genetics, ed 4, St Louis, 2010, Mosby.)
A large number of agents are known to increase the frequency of mutations. These agents are known collectively as mutagens. Radiation, such as that produced by x-rays and nuclear fallout, is an important mutagen and is known to cause cell damage (see Chapter 12). Radiation can fragment the DNA molecule and it can cause chemical reactions that can alter a DNA base. A variety of chemicals also can induce mutations, often because they are chemically similar to DNA bases. Other chemicals mimic the effects of ionizing radiation, and still others interfere with the process of base pairing. Hundreds of chemicals are now known to be mutagenic in humans or laboratory animals, such as nitrogen mustard, vinyl chloride, alkylating agents, formaldehyde, and sodium nitrite. Some of these chemicals, however, are much more potent mutagens than others. Nitrogen mustard, for example, is extremely mutagenic, whereas sodium nitrate is a weak mutagen.
Measurement of the mutation rate in humans is difficult, in part because mutations are very rare events. Current estimates are that the mutation rate in humans is about 10−4 to 10−7 per gene per generation. This rate appears to vary from one gene to another, with greater mutation rates for larger genes. At the nucleotide level, the human mutation rate is approximately 10−8 per nucleotide per generation. Certain DNA sequences have particularly high mutation rates and are known as mutational hot spots. In particular, sequences consisting of a cytosine base followed by a guanine base (CG) are highly susceptible to mutation and are known to account for a disproportionately large percentage of disease-causing mutations.3
From Genes to Proteins
Transcription
Transcription is the process by which RNA is synthesized from a DNA template. The result is the formation of messenger RNA (mRNA) from the base sequence specified by the DNA molecule. Transcription of a gene begins when an enzyme called RNA polymerase binds to a promoter site on the DNA. A promoter site is a sequence of DNA that specifies the beginning of a gene. In addition to RNA polymerase, proteins called transcription factors bind to DNA sequences called transcription factor binding sites near genes to regulate the timing of transcription, as well as the specific tissues in which genes are actively transcribed (e.g., clotting factor VIII primarily in hepatocytes) (Figure 4-5). Transcription factors can either activate or repress the expression of genes. In addition, transcription is sometimes up-regulated by the binding of nearby DNA sequences called enhancers. The RNA polymerase pulls a portion of the DNA strands apart from one another, allowing unattached DNA bases to be exposed. One of the DNA strands then provides the template for the sequence of mRNA nucleotides.
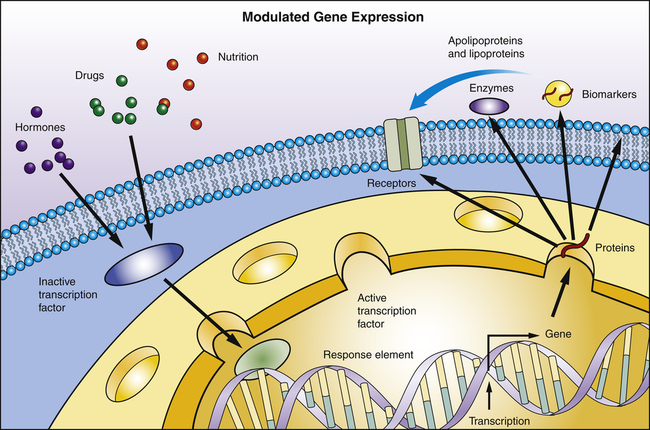
Transcription factors are proteins and bind to specific sites on DNA. They read and interpret the genetic blueprint of the DNA thereby controlling transcription or the flow of genetic information from DNA to mRNA. Transcription factors are essential for gene expression; for example, proteins that function as receptors, enzymes, or biomarkers. Environmental stimuli: for example, hormones involve signaling cascades that can involve transcription factors. Transcription factors can alter gene expression to promote pathophysiology.
The sequence of bases in the mRNA is thus complementary to that of the template strand, and with the exception of the presence of uracil instead of thymine, the mRNA sequence is identical to that of the other DNA strand. Transcription continues until a DNA sequence called a termination sequence is reached. Then the RNA polymerase detaches from the DNA, and the transcribed mRNA is freed to move out of the nucleus and into the cytoplasm. Figure 4-6 summarizes the process of transcription.
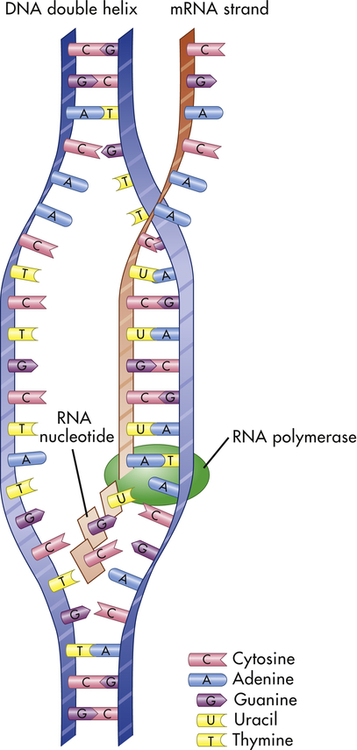
Translation
Translation is the process by which RNA directs the synthesis of a polypeptide (Figure 4-7). However, mRNA cannot code directly for amino acids. Instead, it interacts with transfer RNA (tRNA), a cloverleaf-shaped strand of about 80 nucleotides. The tRNA molecule has a site for the attachment of an amino acid. At the opposite side of the cloverleaf is a sequence of three nucleotides called the anticodon. The anticodon undergoes complementary base pairing with an appropriate codon in the mRNA. The mRNA thus specifies the sequence of amino acids by acting through the tRNA. As each codon is processed, an amino acid is translated by the interaction of mRNA and tRNA, which is aided by cytoplasmic structures called ribosomes. Bonds are formed between adjacent amino acids to make a growing polypeptide. When the ribosome arrives at a termination signal on the mRNA sequence, translation and polypeptide formation cease. The mRNA, ribosome, and polypeptide separate from one another and the polypeptide is released into the cytoplasm to perform its required function.
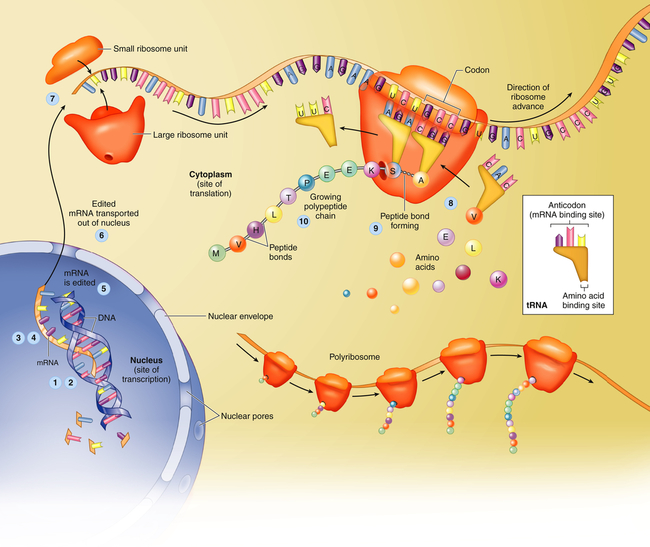
Protein synthesis begins with transcription, a process in which an mRNA molecule forms along one gene sequence of a DNA molecule within the cell’s nucleus (1-3). As it is formed, the mRNA molecule separates from the DNA molecule (4), is edited (5), and leaves the nucleus through the large nuclear pores (6). Outside the nucleus, ribosome subunits attach to the beginning of the mRNA molecule and begin the process of translation (7). In translation, transfer RNA (tRNA) molecules bring specific amino acids—encoded by each mRNA codon—into place at the ribosome site (8). As the amino acids are brought into the proper sequence, they are joined together by peptide bonds (9) to form long strands called polypeptides (10). Several polypeptide chains may be needed to make a complete protein molecule. A, Adenine; C, cytosine; G, guanine; U, uracil. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 8, St Louis, 2013, Mosby.)
Chromosomes
Human cells can be categorized into two types: gametes (sperm and egg cells) and somatic cells, which include all cells other than gametes. Each somatic cell has 46 chromosomes in its nucleus. These are diploid cells, meaning that the chromosomes occur in pairs. Thus each cell actually contains 23 pairs of chromosomes. One member of each pair comes from an individual’s mother, and one comes from the father. New somatic cells are formed through mitosis and cytokinesis, through which the cell nucleus and cytoplasm are replicated. (The division process that creates new copies of somatic cells is described in Chapter 1.) Gametes are haploid cells: they have only one member of each chromosome pair, giving them a total of 23 chromosomes. The process by which these haploid cells are formed from diploid cells is called meiosis (Figure 4-8).
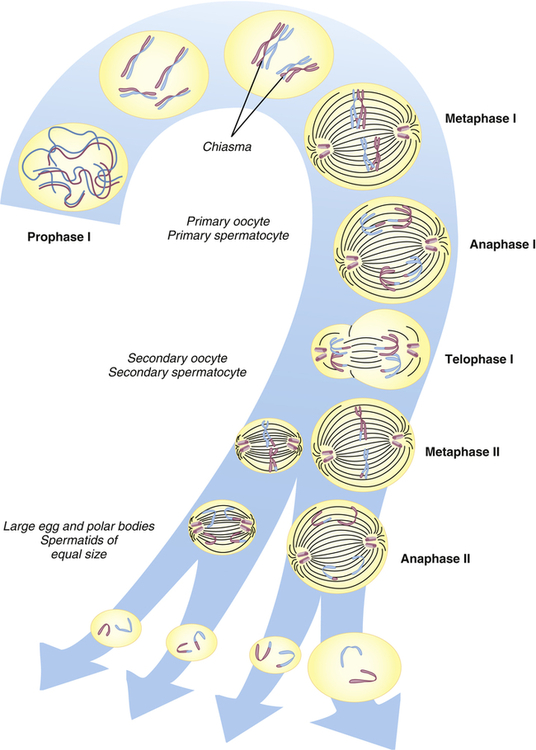
From these stages, haploid gametes are formed from a diploid stem cell. For brevity, prophase II and telophase II are not shown. Note the relationship between meiosis and spermatogenesis and oogenesis. (From Jorde LB et al: Medical genetics, ed 4, St Louis, 2010, Mosby.)
Figure 4-9, A, illustrates a metaphase spread, which is a photograph of the chromosomes as they appear in the nucleus of a somatic cell during metaphase. (Chromosomes are easiest to visualize during this stage of mitosis.) A karyotype is an ordered display of chromosomes. In Figure 4-9, B, the chromosomes are arranged according to size, with the homologous chromosomes paired. The 22 autosomes are numbered according to length, with chromosome 1 as the longest and chromosome 22 as the shortest. Some natural variation in relative chromosome length can be expected from person to person, however, so it is not always possible to distinguish each chromosome by its length. Therefore, the position of the centromere is also used to classify the chromosomes (Figure 4-10).
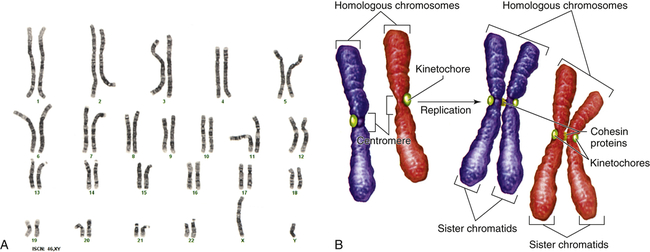
A, Human karyotype. B, Homologous chromosomes and sister chromatids. (A courtesy of the Clinical Cytogenetics Section, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, MD. B from Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
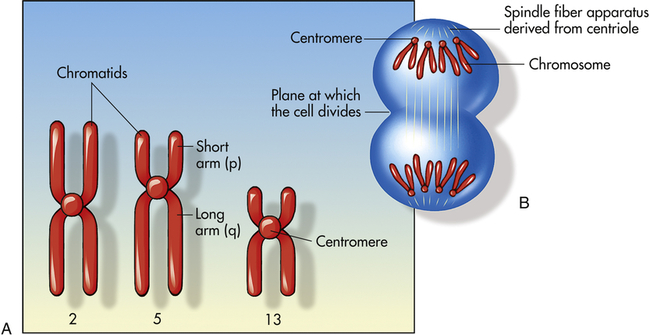
A, Human chromosomes 2, 5, and 13. Each is replicated and consists of two chromatids. Chromosome 2 is a metacentric chromosome because the centromere is close to middle; chromosome 5 is submetacentric because the centromere is set off from the middle; chromosome 13 is acrocentric because the centromere is at or very near the end. B, During mitosis, the centromere divides and chromosomes move to opposite poles of the cell. At the time of centromere division, the chromatids are designated chromosomes.
The chromosomes in Figure 4-9, A, were stained with a substance that binds preferentially to certain areas of chromosomes. The resulting distinctive chromosome bands are evident in various patterns in the different chromosomes so that each chromosome can be distinguished easily. One of the most commonly used stains is Giemsa stain. By using banding techniques, chromosomes can be unambiguously numbered, and individual variation in chromosome composition can be studied. Missing or duplicated portions of chromosomes, which often result in serious diseases, also can be readily identified.
Chromosome Aberrations and Associated Diseases
Chromosome abnormalities are the leading known cause of mental retardation and miscarriage. A major chromosome aberration occurs in more than half of conceptions. Most of these fetuses do not survive to term; in fact, about 50% of all recovered first-trimester spontaneous abortuses have major chromosomal aberrations.4 About 1 in 150 live births has a major diagnosable chromosome abnormality.5
Polyploidy
Cells that have a multiple of the normal number of chromosomes are said to be euploid cells (Greek eu = good or true). Because normal gametes are haploid and most normal somatic cells are diploid, they are both euploid forms. When a euploid cell has more than the diploid number of chromosomes, it is said to be a polyploid cell. Several types of body tissues, including some liver, bronchial, and epithelial tissues, are normally polyploid. A zygote having three copies of each chromosome, rather than the usual two, has a form of polyploidy called triploidy. Tetraploidy, a condition in which euploid cells have 92 chromosomes, also has been observed. Nearly all triploid and tetraploid conceptions are spontaneously aborted or stillborn, and the small proportion that survive to term die shortly after birth. Triploidy and tetraploidy are relatively common conditions at conception, accounting for approximately 10% of all known miscarriages.4
Aneuploidy
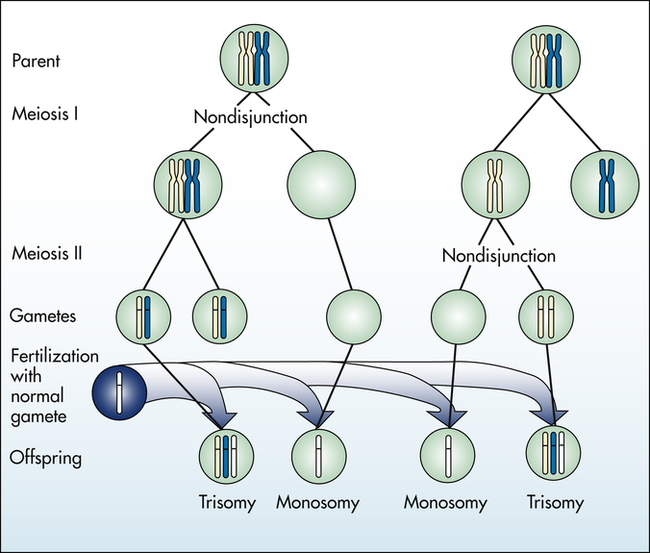
Aneuploidy is usually the result of nondisjunction, an error in which homologous chromosomes or sister chromatids fail to separate normally during meiosis or mitosis (Figure 4-11). Nondisjunction during either stage of meiosis produces some gametes that have two copies of a given chromosome and others that have no copies of the chromosome. When such gametes unite with normal haploid gametes, the resulting zygote is monosomic or trisomic for that chromosome.
Autosomal Aneuploidy
Trisomy can occur for any chromosome at conception, but the only forms seen with an appreciable frequency in live births are trisomies of the thirteenth, eighteenth, or twenty-first chromosome. Fetuses with most other chromosomal trisomies do not survive to term. Trisomy 16, for example, is the most commonly known trisomy among abortuses, but it is not seen in live births.4
The most well-known example of aneuploidy in an autosome is trisomy of the twenty-first chromosome, which causes Down syndrome (named after J. Langdon Down, who first described the disease in 1866). Down syndrome was formerly called mongolism, but this inappropriate term is no longer used. Down syndrome is seen in 1 in 800 live births.4 Individuals with this disease typically have intelligence quotients (IQs) between 25 and 70. The facial appearance is distinctive (Figure 4-12), with a low nasal bridge, epicanthal folds, protruding tongue, and flat, low-set ears. Poor muscle tone (hypotonia) and short stature are both characteristic. Congenital heart defects affect about one third to one half of live-born children with Down syndrome; a reduced ability to fight respiratory tract infections and an increased susceptibility to leukemia also contribute to reduced survival rate. By 40 years of age, individuals with Down syndrome virtually always develop symptoms that are nearly identical to those of Alzheimer disease because one of the genes that can cause Alzheimer disease is located on chromosome 21. About three fourths of fetuses known to have Down syndrome are spontaneously aborted or stillborn. About 20% of infants born with Down syndrome die during their first 10 years of life. For those who survive beyond 10 years, average life expectancy is now about 60 years.
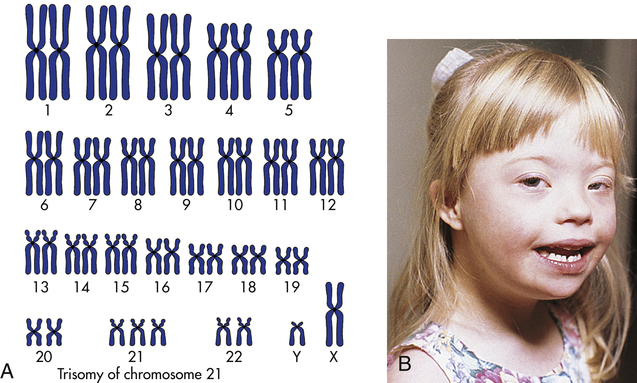
A, The karyotype of Down syndrome consists of 47 chromosomes and shows trisomy 21. B, A child with Down syndrome. (A from Damjanov I: Pathology for the health professions, ed 4, Philadelphia, 2012, Saunders; B courtesy Olney A, MacDonald M, University of Nebraska Medical Center, Omaha.)
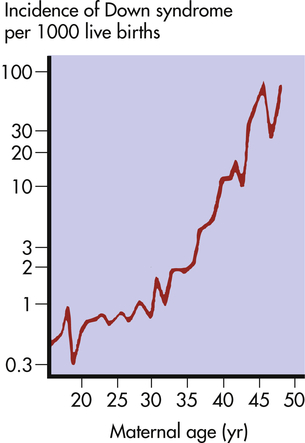
The risk of having a child with Down syndrome increases greatly with maternal age. As Figure 4-13 demonstrates, women younger than 30 years have a risk ranging from about 1 in 1000 births to 1 in 2000 births. The risk begins to rise substantially after 35 years of age, and it reaches 3% to 5% for women older than 45 years of age. This dramatic increase in risk is a consequence of the age of maternal egg cells, which are held in an arrested state of prophase I from the time they are formed in the female embryo until they are shed in ovulation. Thus an egg cell formed by a 45-year-old woman is itself 45 years old. This long suspended state may allow for the accumulation of errors leading to nondisjunction. The risk of Down syndrome, as well as other trisomies, does not appear to increase with paternal age.6
Sex Chromosome Aneuploidy
Among live births, about 1 in 400 males and 1 in 650 females have a form of sex chromosome aneuploidy.7 Because these conditions are generally less severe than autosomal aneuploidies, all forms except complete absence of an X chromosome allow at least some individuals to survive.
One of the most common sex chromosome aneuploidies, affecting about 1 in 1000 newborn females, is trisomy X. Instead of two X chromosomes, these females have three X chromosomes in each cell. Most of them have no overt physical abnormalities, although sterility, menstrual irregularity, or cognitive deficits are sometimes seen. A very small proportion of females have four or even five X chromosomes, and their mental function is more severely compromised. Another sex chromosome aneuploidy is the presence of a single X chromosome and no homologous X or Y chromosome, resulting in a total of 45 chromosomes. The karyotype is designated 45,X, and it causes a set of symptoms known as Turner syndrome (Figure 4-14). Because they have no Y chromosome, only females are affected by Turner syndrome. They are usually sterile, however, and have gonadal streaks rather than ovaries. These streaks of connective tissue are susceptible to cancer in mosaics who have some cells containing a Y chromosome. Other features of the disorder include short stature, webbing of the neck in about half of cases, widely spaced nipples, coarctation (narrowing) of the aorta (in 15% to 20% of cases), edema of the feet in newborns, and sparse body hair. Their IQs are typically in the normal range, although they often have some impairment of spatial and mathematical reasoning ability. About three fourths of recognized 45,X conceptions inherit their X chromosome from the mother. Thus most cases are caused by a loss of the paternal X chromosome.
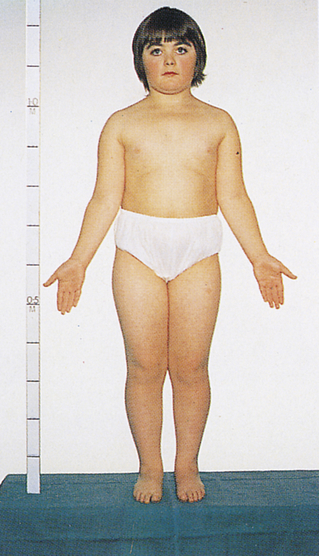
A sex chromosome is missing, and the person’s chromosomes are 45,X. Characteristic signs are short stature, female genitalia abnormality, webbed neck, shieldlike chest with underdeveloped breasts and widely spaced nipples, and imperfectly developed ovaries. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 8, St Louis, 2013, Mosby.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


