Chapter 28
Alterations of Erythrocyte Function
Neal S. Rote and Kathryn L. McCance

Anemia
Classification
Anemias are classified by their causes (e.g., anemia of chronic disease) or by changes that affect the size, shape, or hemoglobin content of the erythrocyte (Box 28-1). The most common classification is based on changes that affect the erythrocyte’s size or hemoglobin content (Table 28-1). The terminology reflects these characteristics; terms that end in “-cytic” refer to cell size, whereas “-chromic” refers to hemoglobin content (Table 28-2). Additional descriptors of erythrocytes associated with some anemias include anisocytosis (assuming various sizes) or poikilocytosis (assuming various shapes) (Figure 28-1).
TABLE 28-1
MORPHOLOGIC CLASSIFICATION OF ANEMIAS
| MORPHOLOGY OF REMAINING ERYTHROCYTES | NAME AND MECHANISM OF ANEMIA | PRIMARY CAUSE |
| Macrocytic-normochromic anemia: large, abnormally shaped erythrocytes but normal hemoglobin concentrations | Pernicious anemia: lack of vitamin B12 (cobalamin) for erythropoiesis; abnormal deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis in the erythroblast; premature cell death | Congenital or acquired deficiency of intrinsic factor (IF); genetic disorder of DNA synthesis |
| Folate deficiency anemia: lack of folate for erythropoiesis; premature cell death | Dietary folate deficiency | |
| Microcytic-hypochromic anemia: small, abnormally shaped erythrocytes and reduced hemoglobin concentration | Iron deficiency anemia: lack of iron for hemoglobin production; insufficient hemoglobin | Chronic blood loss; dietary iron deficiency; disruption of iron metabolism or iron cycle (see Chapter 27) |
| Sideroblastic anemia: dysfunctional iron uptake by erythroblasts and defective porphyrin and heme synthesis | Congenital dysfunction of iron metabolism in erythroblasts; acquired dysfunction of iron metabolism as a result of drugs or toxins | |
| Thalassemia: impaired synthesis of α- or β-chain of hemoglobin A; phagocytosis of abnormal erythroblasts in the marrow | Congenital genetic defect of globin synthesis | |
| Normocytic-normochromic anemia: normal size, normal hemoglobin concentration | Aplastic anemia: insufficient erythropoiesis | Depressed stem cell proliferation resulting in bone marrow aplasia |
| Posthemorrhagic anemia: blood loss | Acute or chronic hemorrhage that stimulates increased erythropoiesis, which eventually depletes body iron | |
| Hemolytic anemia: premature destruction (lysis) of mature erythrocytes in the circulation | Increased fragility of erythrocytes | |
| Sickle cell anemia: abnormal hemoglobin synthesis, abnormal cell shape with susceptibility to damage, lysis, and phagocytosis | Congenital dysfunction of hemoglobin synthesis | |
| Anemia of chronic disease: abnormally increased demand for new erythrocytes | Chronic infection or inflammation; malignancy |
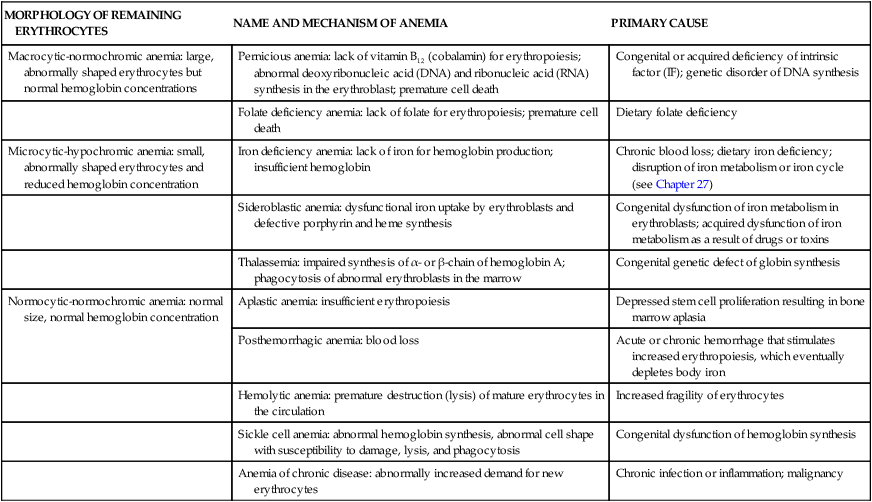
TABLE 28-2
TERMS USED IN ASSESSMENT OF ERYTHROCYTES
| ERYTHROCYTE VOLUME | HEMOGLOBIN CONTENT | |
| Normal | Normocytic | Normochromic |
| Increased | Macrocytic (higher mean corpuscular volume [MCV]) | Hyperchromic (higher mean corpuscular hemoglobin concentration [MCHC]) |
| Decreased | Microcytic (lower MCV) | Hypochromic (lower MCHC) |
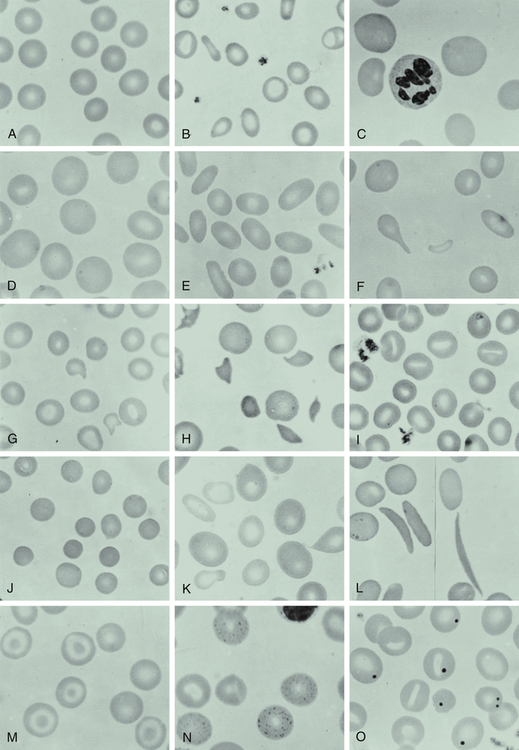
A, Normal blood smear. B, Microcytic-hypochromic anemia (iron deficiency). C, Macrocytic anemia (pernicious anemia). D, Macrocytic anemia in pregnancy. E, Hereditary elliptocytosis. F, Myelofibrosis (teardrop). G, Hemolytic anemia associated with prosthetic heart valve. H, Microangiopathic anemia. I, Stomatocytes. J, Spherocytes (hereditary spherocytosis). K, Sideroblastic anemia; note the double population of red blood cells. L, Sickle cell anemia. M, Target cells (after splenectomy). N, Basophil stippling in case of unexplained anemia. O, Howell-Jolly bodies (after splenectomy). (From Wintrobe MM et al: Clinical hematology, ed 8, Philadelphia, 1981, Lea & Febiger.)
Clinical Manifestations
The fundamental physiologic manifestation of anemia is a reduced oxygen-carrying capacity of the blood resulting in tissue hypoxia. Symptoms of anemia vary, depending on the body’s ability to compensate for reduced oxygen-carrying capacity (Figure 28-2). Anemia that is mild and develops gradually is usually easier to compensate and may cause problems for the individual only during physical exertion. As the reduction in red blood cells (RBCs) continues, symptoms become more pronounced and alterations of specific organs and compensatory effects become more apparent. Compensation generally involves the cardiovascular, respiratory, and hematologic systems. (Hematologic findings associated with various anemias are listed in Table 28-3 and progression and manifestations of anemias are shown in Figure 28-2.)
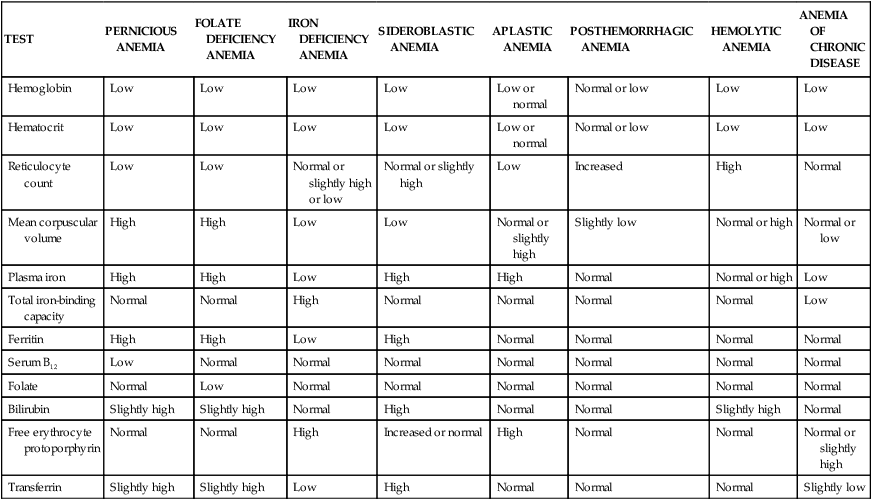
TABLE 28-3
LABORATORY FINDINGS FOR VARIOUS ANEMIAS
| TEST | PERNICIOUS ANEMIA | FOLATE DEFICIENCY ANEMIA | IRON DEFICIENCY ANEMIA | SIDEROBLASTIC ANEMIA | APLASTIC ANEMIA | POSTHEMORRHAGIC ANEMIA | HEMOLYTIC ANEMIA | ANEMIA OF CHRONIC DISEASE |
| Hemoglobin | Low | Low | Low | Low | Low or normal | Normal or low | Low | Low |
| Hematocrit | Low | Low | Low | Low | Low or normal | Normal or low | Low | Low |
| Reticulocyte count | Low | Low | Normal or slightly high or low | Normal or slightly high | Low | Increased | High | Normal |
| Mean corpuscular volume | High | High | Low | Low | Normal or slightly high | Slightly low | Normal or high | Normal or low |
| Plasma iron | High | High | Low | High | High | Normal | Normal or high | Low |
| Total iron-binding capacity | Normal | Normal | High | Normal | Normal | Normal | Normal | Low |
| Ferritin | High | High | Low | High | Normal | Normal | Normal | Normal |
| Serum B12 | Low | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Folate | Normal | Low | Normal | Normal | Normal | Normal | Normal | Normal |
| Bilirubin | Slightly high | Slightly high | Normal | High | Normal | Normal | Slightly high | Normal |
| Free erythrocyte protoporphyrin | Normal | Normal | High | Increased or normal | High | Normal | Normal | Normal or slightly high |
| Transferrin | Slightly high | Slightly high | Low | High | Normal | Normal | Normal | Slightly low |
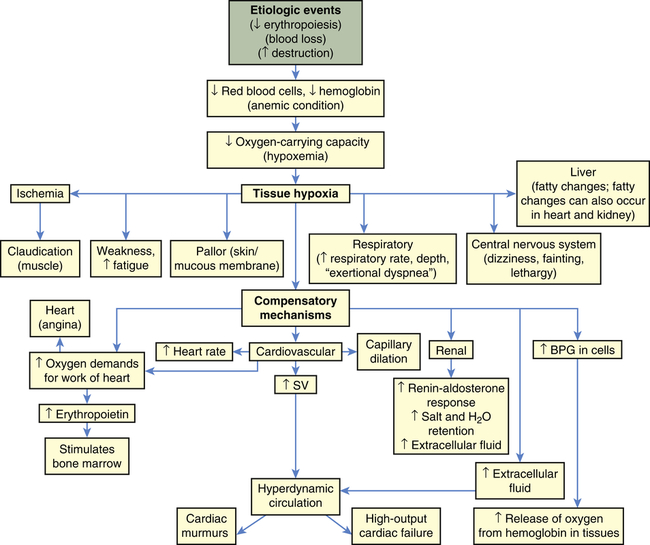
Tissue hypoxia creates additional demands and compensatory actions on the pulmonary and hematologic systems. The rate and depth of breathing increase in an attempt to increase the availability of oxygen. These demands are accompanied by an increase in the release of oxygen from hemoglobin. (Mechanisms of oxygen transport and release by hemoglobin are described in Chapter 27.) All of these compensatory mechanisms may cause individuals to experience shortness of breath (dyspnea); a rapid, pounding heartbeat (palpitations); dizziness; and fatigue. In mild, chronic conditions, these symptoms might be present only when demand for oxygen is increased (e.g., during physical exertion), but in severe conditions they may be experienced at rest.
Macrocytic-Normochromic Anemias
The macrocytic (megaloblastic) anemias are characterized by unusually large stem cells (megaloblasts) in the marrow that mature into erythrocytes that are unusually large in size (macrocytes), thickness, and volume.1 The hemoglobin content is normal (normochromic). These anemias are the result of defective erythrocyte DNA synthesis, commonly caused by deficiencies of vitamin B12 (cobalamin) or folate (folic acid), coenzymes that are required for nuclear maturation and DNA synthesis. These defective erythrocytes die prematurely, which decreases their numbers in the circulation, causing anemia.
Premature death of damaged erythrocytes, eryptosis, is a common mechanism of cellular loss in individuals with anemias secondary to deficiencies of iron, infections (e.g., malaria, mycoplasma), chronic diseases (e.g., diabetes, renal disease), genetic diseases (e.g., beta-thalassemia, glucose-6-phosphate dehydrogenase [G6PD] deficiency, sickle-cell trait), and myelodysplastic syndrome.2 The process is similar to the removal of old or senescent erythrocytes (see Chapter 27), but is triggered by erythrocyte damage before the cell’s normal life span. Damaged erythrocytes undergo cell shrinkage, membrane changes (blebbing), and rearrangement of plasma membrane phospholipid distribution with efflux of phosphatidylserine (PS). Macrophages have receptors that recognize surface PS and remove the damaged erythrocytes from the circulation. The erythrocyte’s life span may be decreased by as much as 50%.3
Pernicious Anemia
Pernicious anemia (PA), the most common type of megaloblastic anemia, is caused by vitamin B12 deficiency, which is often associated with the end stage of type A chronic atrophic (congenital or autoimmune) gastritis (see Figure 28-1, C; Figure 28-3).4 Pernicious means highly injurious or destructive and reflects the fact that this condition was once fatal. It most commonly affects individuals older than the age of 30 (60 being the median age of diagnosis) who are of Northern European descent, primarily those of Scandinavian, English, and Irish descent, and PA is less common in individuals of Greek or Italian origin, Recently, PA has also been reported in blacks and Hispanics. Females are more prone to develop PA, with black females having an earlier onset.
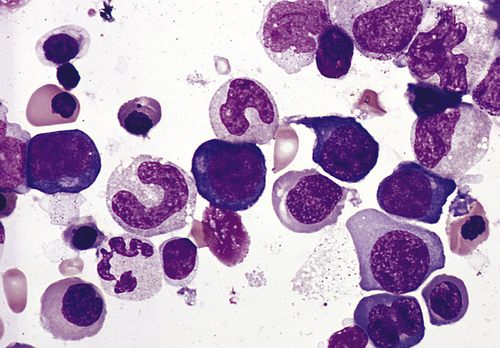
Bone marrow aspirate smear from an individual with megaloblastic red blood cell precursors and giant metamyelocytes. The chromatin in the red blood cell nuclei is more dispersed than that in normal red blood cell precursors at comparable stages of maturation; the giant metamyelocytes have dispersed nuclear chromatin in contrast to a normal metamyelocyte, which has condensed chromatin (Wright-Giemsa stain). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Pathophysiology
Deficiency in IF secretion may be congenital or, more often, an autoimmune process directed against gastric parietal cells. Congenital IF deficiency is a genetic disorder that demonstrates an autosomal recessive inheritance pattern.5 The autoimmune form of the disease also has a genetic component, as do most autoimmune diseases (see Chapter 9). Family clusters have been identified; 20% to 30% of individuals related to persons with PA also have PA. These relatives, particularly first-degree female relatives, also demonstrate a higher frequency of the presence of gastric autoantibodies. PA is also frequently a component of autoimmune polyendocrinopathy, which is a cluster of autoimmune diseases of endocrine organs (e.g., chronic autoimmune thyroiditis [Hashimoto thyroiditis], type 1 diabetes mellitus, Addison disease, primary hypoparathyroidism, Graves disease, and myasthenia gravis) that frequently present as comorbidities. Autoimmune thyroiditis and type 1 diabetes mellitus, in particular, are associated with PA.
Most cases of PA result from an autoimmune gastritis (type A chronic gastritis) in which gastric atrophy results from destruction of parietal and zymogenic cells. Individuals with PA commonly have autoantibodies against the gastric H+-K+ ATPase, which is the major protein constituent of parietal cell membranes. Early in the disease process the gastric submucosa becomes infiltrated with inflammatory cells, including CD4 lymphocytes, eventually extending into the lamina propria and causing degeneration of the parietal and zymogenic cells. The parietal and zymogenic cells are destroyed and replaced by mucous-containing cells (intestinal metaplasia). Gastric mucosal atrophy, in which gastric parietal cells are destroyed, results in a deficiency of all secretions of the stomach—hydrochloric acid, pepsin, and IF. A direct correlation exists between the severity of the gastric lesion and the degree of malabsorption of vitamin B12.6 Additionally, autoantibodies against IF prevent the formation of the B12-IF complex. Thus, PA is secondary to autoimmune destruction of parietal cells, thus diminishing the production of IF, and the presence of autoantibodies that neutralize the capacity of remaining IF to transport vitamin B12.
Initiation of the autoimmune process may be secondary to a past infection with Helicobacter pylori.7 Although active infection with H. pylori is rare in individuals with PA, more than half of these individuals possess circulating antibodies against this microorganism, suggesting a history of infection. The current opinion is that in genetically prone individuals, antigens expressed by H. pylori mimic the parietal cell H+-K+ ATPase, resulting in production of an antibody that binds and damages the parietal cell (see Chapter 9 for a discussion of antigenic mimicry and autoimmune disease).
Evaluation and Treatment
Diagnosis of PA is based on a variety of tests (see Table 28-3), which include blood tests, bone marrow aspiration, serologic studies, gastric biopsy, and clinical manifestations. A good test for PA was the Schilling test (no longer offered in most laboratories), which indirectly evaluated vitamin B12 absorption by administering radioactive B12 and measuring excretion in the urine. Low urinary excretion was significant for PA.
Serologic studies, however, have replaced the Schilling test for diagnosing PA. Measuring methylmalonic acid and homocysteine levels, which are elevated early in PA, is more sensitive. The presence of circulating antibodies against parietal cells and intrinsic factor is also useful in diagnosis.8 Gastric biopsy reveals total achlorhydria (absence of hydrochloric acid), which is diagnostic for PA because it occurs only in the presence of this gastric lesion.
Folate Deficiency Anemia
Pathophysiology
Impaired DNA synthesis secondary to a folate deficiency results in megaloblastic cells with clumped nuclear chromatin. Anemia may result from apoptosis of erythroblasts in the late stages of erythropoiesis. In addition to anemia, folate deficiency in pregnant women is associated with neural tube defects of the fetus. Folate is necessary for the reduction of circulating levels of homocysteine, a risk factor for the development of atherosclerosis (see Chapter 32); thus a folate deficiency increases the risk for developing coronary artery disease. A deficiency of folate also is implicated in the development of cancers, specifically colorectal cancers.
Microcytic-Hypochromic Anemias
The microcytic-hypochromic anemias are characterized by abnormally small erythrocytes that contain abnormally reduced amounts of hemoglobin (see Figure 28-1, B). Hypochromia occurs even in cells of normal size.
Microcytic-hypochromic anemia can result from (1) disorders of iron metabolism, (2) disorders of porphyrin and heme synthesis, or (3) disorders of globin synthesis. Specific disorders include iron deficiency anemia, sideroblastic anemia, and thalassemia (thalassemia is discussed in Chapter 30).
Iron Deficiency Anemia
Children in developing countries often are affected by chronic parasite infestations that result in intestinal blood and iron loss that outpaces dietary intake.9 Treatment of helminth infections results in an improvement in the anemia as well as in appetite and growth. Iron deficiency also occurs in individuals with lead poisoning. Treatment of the iron deficiency is associated with a decrease in lead levels.
Females have a higher incidence of hypoferremia (13.9%) than do males (8.3%), as well as IDA— 4% to 6% in females and 4% in males. The incidence peaks in females during their reproductive years and decreases after menopause. Those at highest risk are black females living in urban poverty.10 Males have a higher incidence during childhood and adolescence, a decrease occurring during young adulthood, and an upswing during late adulthood. In the United States, 720,000 children (9%) ages 1 to 2 years are estimated to be iron deficient, of whom 240,000 (3%) are anemic, which may be a result of increased iron requirements with growth. An increased prevalence of iron deficiency has been observed in overweight children.
Pathophysiology
The most common cause of IDA in developed countries is pregnancy and chronic blood loss.11 Blood loss of 2 to 4 ml/day (1 to 2 mg of iron) is sufficient to cause iron deficiency and may result from erosive esophagitis, gastric and duodenal ulcers, colon adenomas, or cancers. H. pylori infections also have been found to cause IDA of unknown origin, although H. pylori impairs iron uptake. In females, menorrhagia (excessive bleeding during menstruation) is a common cause of primary IDA. Other causes of IDA for both genders are (1) use of medications that cause gastrointestinal bleeding (such as aspirin or nonsteroidal anti-inflammatory drugs [NSAIDs]); (2) surgical procedures that decrease stomach acidity, intestinal transit time, and absorption (e.g., gastric bypass); (3) insufficient dietary intake of iron; and (4) eating disorders, such as pica, which is the craving and eating of nonnutritional substances, such as dirt, chalk, and paper.
Iron in the form of hemoglobin is in constant demand by the body. Iron is recyclable; therefore, the body maintains a balance between iron that is contained in hemoglobin and iron that is in storage and available for future hemoglobin synthesis (see Chapter 27). Blood loss disrupts this balance by creating a need for more iron, thus depleting the iron stores more rapidly to replace the iron lost from bleeding.
Clinical Manifestations
Symptoms of IDA begin gradually, and individuals usually do not seek medical attention until hemoglobin levels have decreased to about 7 to 8 g/dl. Early symptoms are nonspecific and include fatigue, weakness, shortness of breath, and pale earlobes, palms, and conjunctivae (Figure 28-4, A).
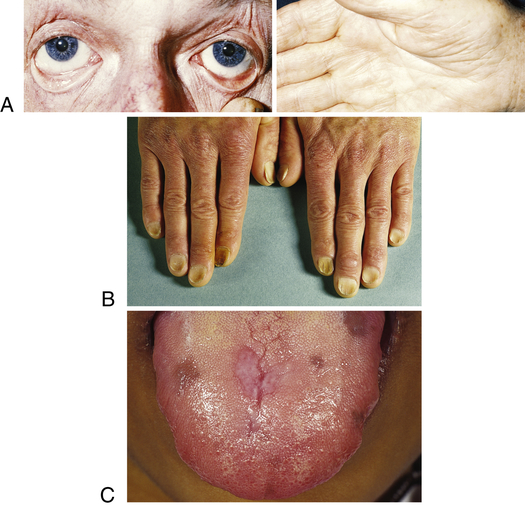
A, Pallor and iron deficiency. Pallor of the skin, mucous membranes, and palmar creases in an individual with a hemoglobin level of 9 g/dl. Palmar creases become as pale as the surrounding skin when the hemoglobin level approaches 7 g/dl. B, Koilonychia. The nails are concave, ridged, and brittle. C, Glossitis. Tongue of individual with iron deficiency anemia has bald, fissured appearance caused by loss of papillae and flattening. (From Hoffbrand AV, Pettit JE, Vyas P: Color atlas of clinical hematology, ed 4, London, 2009, Mosby; B courtesy Dr. S.M. Knowles.)
As the condition progresses and becomes more severe, structural and functional changes occur in epithelial tissue (see Figure 28-4). The fingernails become brittle, thin, coarsely ridged, and “spoon-shaped” or concave (koilonychia) as a result of impaired capillary circulation (Figure 28-4, B). IDA also is associated with unexplained burning mouth syndrome, as was discussed for folate deficiency. Tongue papillae atrophy and cause soreness along with redness and burning (glossitis) (Figure 28-4, C). The degree of pain experienced is directly associated with the amount of iron deficiency, and these changes can be reversed within 1 to 2 weeks of iron replacement therapy. Individuals also experience dryness and soreness in the epithelium at the corners of the mouth, known as angular stomatitis. Difficulty in swallowing is associated with an esophageal “web,” a thin, concentric, smooth extension of normal esophageal tissue consisting of mucosa and submucosa at the juncture between the hypopharynx and esophagus. The duration of iron deficiency required for web formation is uncertain. Dysphagia also is exacerbated by hyposalivation. The pathophysiology associated with these epithelial lesions is not well understood, but the lesions have the potential to become cancerous.
Evaluation and Treatment
Initial evaluation is based on clinical symptoms and decreased levels of hemoglobin and hematocrit. Additional measurements, however, are needed to determine the cause of the anemia (see Table 28-3). Iron stores may be measured directly by bone marrow biopsy and iron staining or indirectly by laboratory tests for serum ferritin, transferrin saturation, or total iron-binding capacity. Serum ferritin is a widely accepted and available measurement of iron status that has been used for the past 25 years; 1 mcg/L serum ferritin corresponds to 8 to 10 mg or 120 mcg of storage iron/kg body weight. A limitation on interpretation of serum ferritin levels is that values may be elevated independently of iron status during acute or chronic inflammation, malignancy, liver disease, or alcoholism. A sensitive indicator of heme synthesis is the amount of free erythrocyte protoporphyrin (FEP) within erythrocytes. A test that determines the concentration of soluble fragment transferrin receptor differentiates primary IDA from IDA that is associated with chronic disease.
The first step in treatment of IDA is to identify and eliminate sources of blood loss.12 With ongoing bleeding, any replacement therapy is likely to be ineffective. Iron replacement therapy is required and very effective. Initial doses are 150 to 200 mg/day. Hematocrit levels should improve within 1 to 2 months of therapy; however, the serum ferritin level is a more precise measurement of improvement and total body stores of iron. Once the serum ferritin level reaches 50 mcg/L, adequate replacement of iron has occurred. A rapid decrease in fatigue, lethargy, and other associated symptoms is generally seen within the first month of therapy. Replacement therapy usually continues for 6 to 12 months after the bleeding has stopped but may continue for as long as 24 months. Menstruating females may need daily oral iron replacement therapy (325 mg/day) until menopause.
Sideroblastic Anemia
Sideroblastic anemias (SAs) are a heterogeneous group of disorders characterized by anemia of varying severity caused by a defect in mitochondrial heme synthesis.13 SA is characterized by the presence of ringed sideroblasts within the bone marrow. Ringed sideroblasts are erythroblasts that contain iron-laden mitochondria arranged in a perinuclear collar around one third or more of the nucleus (see Figure 28-1, K).14 Individuals with SA also have increased levels of iron in their tissue. The blood contains hypochromic erythrocytes, either microcytic or macrocytic depending on the form of the disease.
Pathophysiology
SAs have multiple etiologies but all share the commonality of altered heme synthesis in the erythroid cells in bone marrow. Mitochondrial aminolevulinic acid (ALA) synthase uses glycine to convert succinyl CoA into ALA.15 ALA undergoes further enzymatic modification in the cytoplasm to the porphyrin structure, becoming coproporphyrinogen III, which reenters the mitochondria. Within the mitochondria the molecule is progressively converted to protophorphyrin IX, which has ferrous iron (Fe2+) inserted by the enzyme ferrochelatase. Disruptions to this pathway lead to the accumulation of iron in the mitochondria and the characteristic sideroblasts.
Hereditary sideroblastic anemia is rare and occurs almost exclusively in males, suggesting a predominant recessive X-linked transmission. Hereditary SA (X-linked sideroblastic anemia [XLSA]) has been linked to missense mutations in the erythroid-specific ALAS-E gene Xp11.21.16 More than 25 missense mutations have been identified. ALAS is the first and rate-limiting enzyme in the heme biosynthesis pathway, and mutations lead to reduced synthesis of protoporphyrin IX and the characteristic accumulation of iron in the erythrocyte. An occasional autosomal recessive transmission affecting females occurs with mitochondrial mutations and deficiencies of ferrochelatase. Other genetic, chromosomal, or enzyme dysfunctions also have been associated with hereditary SA. The anemia of hereditary SA is usually present in infancy or childhood, but may remain undetected until midlife. In some instances, other symptoms (e.g., diabetes or cardiac failure resulting from tissue iron overload) may be the first manifestation of SA. Differentiation of SA from idiopathic hemochromatosis needs to be confirmed because both are characterized by tissue iron deposition.
The leading known cause of primary ASA, myelodysplastic syndrome (MDS), is a group of disorders of hematopoietic stem cells, with all three stem cell lines demonstrating dysplastic characteristics.17 Initially, all ASAs associated with myelodysplastic syndrome were considered to be one and the same and identified as refractory anemia with ringed sideroblasts. This classification proved unsatisfactory because different outcomes were observed in individuals who had the same apparent disease. Further investigations discovered morphologic and chromosomal characteristics that predicted different clinical courses. Two subsets of myelodysplastic ringed sideroblasts were identified based on the cell lines that were affected. In one subset, dysplastic features were limited to the erythroid line and it was classified as pure SA. Individuals with pure SA require transfusions, which may produce iron overload.18 With adequate chelation therapy, they are able to survive and thrive for many years. A significant outcome of this condition is the rare occurrence of conversion to leukemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


