Chapter 22
Alterations of Hormonal Regulation
Valentina L. Brashers, Robert E. Jones and Sue E. Huether

Mechanisms of Hormonal Alterations
Significantly elevated or depressed hormone levels may result from a variety of causes (Table 22-1). Dysfunction of an endocrine gland may involve the gland’s failure to produce adequate amounts of biologically free or active hormone forms. This failure may occur when the secretory cells are unable to produce or obtain an adequate quantity of required hormone precursors or when they are unable to convert the precursors to the active hormone. A gland also may synthesize or release excessive amounts of hormone. For example, feedback systems that recognize the need for a particular hormone may fail to function properly or may respond to inappropriate signals. Once hormones are released into the circulation, they may be degraded at an altered rate or they may be inactivated by antibodies before reaching the target cell. Ectopic sources of hormones (hormones produced by nonendocrine tissues) may result also in abnormally elevated hormone levels without benefit of the normal feedback system for hormone control. In these cases the ectopic hormone production is said to be autonomous.
TABLE 22-1
MECHANISMS OF HORMONE ALTERATIONS
| INAPPROPRIATE AMOUNTS OF HORMONE DELIVERED TO TARGET CELL | INAPPROPRIATE RESPONSE BY TARGET CELL |
| Inadequate hormone synthesis Inadequate quantity of hormone precursors Secretory cell unable to convert precursors to active hormone Failure of feedback systems Do not recognize positive feedback leading to inadequate hormone synthesis Do not recognize negative feedback leading to excessive hormone synthesis Hormones inactive Inadequate biologically free hormone Hormone degraded at an altered rate Circulating inhibitors Dysfunctional delivery system Inadequate blood supply Inadequate carrier proteins Ectopic production of hormones | Cell surface receptor–associated disorders Decrease in the number of receptors Impaired receptor function (altered affinity for hormones) Presence of antibodies against specific receptors Unusual expression of receptor function Intracellular disorders Acquired defects in postreceptor signaling cascades Inadequate synthesis of a second messenger Intracellular enzymes or proteins are altered Alterations in nuclear co-regulators Altered protein synthesis |
Target cells may fail to respond to its hormone (hormone insensitivity) (see Table 22-1). The general types of abnormal target cell responses currently recognized are receptor-associated disorders and intracellular disorders. Receptor-associated disorders have been identified primarily in water-soluble hormones, such as insulin. These types of disorders usually involve one of the following: (1) a decrease in the number of receptors, leading to decreased or defective hormone-receptor binding; (2) impaired receptor function, resulting in insensitivity to the hormone; (3) presence of antibodies against specific receptors that either reduce available binding sites or mimic hormone action; or (4) unusual expression of receptor function, as occurs in some tumor cells.
Intracellular disorders may involve acquired defects in postreceptor signaling cascades or inadequate synthesis of a second messenger, such as cyclic adenosine monophosphate (cAMP), needed to transduce the hormonal signal into intracellular events. The target cell for water-soluble hormones may have a faulty response to hormone-receptor binding and thus fail to generate the required second messenger, or the cell also may have an abnormal response to the second messenger if levels of intracellular enzymes or proteins are altered. (Second messengers for various hormones are listed in Table 21-3.) Both of these pathogenic mechanisms result in failure of the target cell to express the usual hormonal effect. Pathogenic mechanisms affecting target cell response for lipid-soluble hormones, such as thyroid hormone or glucocorticoids, are recognized less often than those affecting the water-soluble hormones. These hormone-resistant states have been generally linked to mutations in the nuclear receptor for the hormone. The number of receptors may be decreased, or those receptors may have an altered affinity or selectivity for hormones.1 In other cases, hormone responsiveness may be linked to alterations in nuclear co-regulators, which are proteins (such as cAMP response element–binding protein [CREB], see Chapter 21 p. 694) that facilitate or inhibit the transcription of the target gene.2 Alterations in generation of new messenger ribonucleic acid (mRNA) or absence of substrates for new protein synthesis also may occur, resulting in altered target cell response.
Alterations of the Hypothalamic-Pituitary System
Perhaps the most common cause of apparent hypothalamic dysfunction is interruption of the pituitary stalk, which can be caused by destructive lesions, rupture after head injury, surgical transection, or tumor. The absence of hypothalamic releasing or inhibiting hormones (Figure 22-1) causes a variety of manifestations that present clinically as pituitary disease. For example, if there is an absence of gonadotropin-releasing hormone (GnRH) from the hypothalamus, then there is a lack of stimulation of gonadotropin follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary; thus, menses cease in women and spermatogenesis is impaired in men.
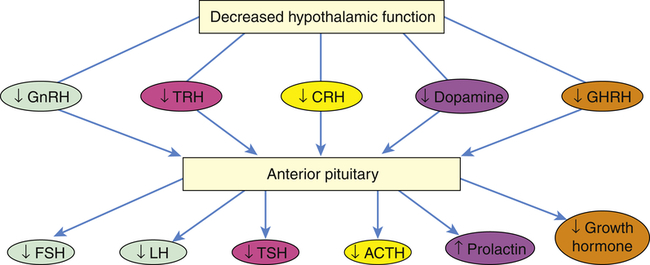
ACTH, Adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; FSH, follicle-stimulating hormone; GHRH, growth hormone–releasing hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
Diseases of the Posterior Pituitary
Syndrome of Inappropriate Antidiuretic Hormone Secretion
Syndrome of inappropriate antidiuretic hormone (SIADH) secretion is characterized by high levels of ADH in the absence of normal physiologic stimuli for its release. SIADH can complicate malignancies, pulmonary disorders, central nervous system disorders, surgical procedures, and the use of certain medications.3 SIADH is associated with ectopic secretion of ADH by several types of tumor cells. Tumors that have been reported in association with SIADH include small cell carcinoma of the lung, duodenum, stomach, and pancreas; cancers of the bladder, prostate, and endometrium; lymphomas; and sarcomas. Pulmonary disorders associated with SIADH include pneumonia (e.g., tuberculosis), asthma, cystic fibrosis, and respiratory failure requiring mechanical ventilation. Central nervous system disorders that may cause SIADH include encephalitis, meningitis, intracranial hemorrhage, tumors, and trauma. A nephrogenic form of SIADH has recently been described. In these individuals, mutations in arginine vasopressin (AVP) genes lead to chronic activation of tubular V2 receptor and resulting excessive free water reabsorption.4 Any surgery can result in postoperative fluid volume shifts and transient SIADH for as long as 5 to 7 days after surgery. The precise mechanism is uncertain but is likely related to fluid and volume changes following surgery, the amount and type of intravenous fluids given, and the use of narcotic analgesics. Transient SIADH is especially common after pituitary surgery because stored ADH is released in an unregulated fashion. Medications are an important cause of SIADH, especially in older adults. These include antidepressants, antipsychotics, narcotics, general anesthetics, chemotherapeutic agents, nonsteroidal anti-inflammatory drugs, quinolone antibiotics, and synthetic ADH analogs.3 These drugs serve either to simulate ADH release, to enhance the physiologic effects of ADH, or to have a biologic action similar to that of ADH.
Pathophysiology
The pathophysiologic features of SIADH are the result of enhanced renal water retention. Water retention results from the action of ADH on renal collecting ducts, where it increases their permeability to water, thus increasing water reabsorption by the kidneys. (Renal function is discussed in Chapter 37.) This results in an expansion of extracellular fluid volume that leads to dilutional hyponatremia (low serum sodium concentration), hypoosmolarity, and urine that is inappropriately concentrated with respect to serum osmolarity.
Evaluation and Treatment
A diagnosis of SIADH requires documentation of the following manifestations: (1) serum hypoosmolality (<280 mOsm/kg) and hyponatremia (serum sodium level <135 mEq/L); (2) urine hyperosmolarity (i.e., the osmolality of the urine is always higher than the concurrent serum osmolality); (3) urine sodium excretion that matches sodium intake; (4) normal renal, adrenal, and thyroid function; and (5) absence of conditions that can alter volume status (e.g., recent diuretic use, heart failure, hypervolemia from any cause, or renal insufficiency).3 Individuals with neurologic injury also may develop hyponatremia caused by cerebral salt wasting syndrome. This can be differentiated from SIADH because cerebral salt wasting is characterized by hypovolemia and weight loss. In addition, urine sodium levels are elevated.5
The treatment of SIADH involves the correction of the underlying causal problems; emergency correction of severe hyponatremia by administration of hypertonic saline; and, most importantly, fluid restriction to 800 to 1000 ml/day. Careful monitoring is important. Resolution usually occurs within 3 days, with a 2- to 3-kg weight loss resulting from enhanced free water clearance. If hyponatremia is corrected too rapidly, a severe neurologic syndrome called central pontine myelinolysis can ensue. No drug therapy is available to suppress ectopically produced ADH; however, demeclocycline, which causes the renal tubules to develop resistance to ADH, may be used to treat resistant or chronic SIADH. An ADH receptor agonist, conivaptan, has been approved for the treatment of hospitalized individuals with hyponatremia caused by ADH excess.6 An oral form of ADH receptor antagonists has been developed.7
Diabetes Insipidus
Neurogenic DI is the form encountered most often in clinical practice and is caused by insufficient amounts of ADH. It occurs when any organic lesion of the hypothalamus, pituitary stalk, or posterior pituitary interferes with ADH synthesis, transport, or release.3 Causative lesions include primary or secondary brain tumors, aneurysms, thrombosis, infections, and immunologic disorders. DI is a well-recognized complication of closed-head trauma and of pituitary surgery in which DI can be transient or permanent.8 Neurogenic DI can be a complication of pregnancy. In rare cases, it also can be caused by hereditary disorders that affect ADH genes or result in structural changes in the pituitary gland, such as septo-optic dysplasia (absence of the septum pellucidum in the brain and underdevelopment of the optic nerve).9
Nephrogenic DI is associated with an insensitivity of the renal collecting tubules to ADH. The nephrogenic form of DI can be genetic or acquired.3 This form of DI is often idiopathic, although several genetic abnormalities that affect the vasopressin receptor have been noted. One of the best described is a mutation in the gene that codes for aquaporin-2, which is one of the four water transport channels in the renal tubule.9
Dipsogenic DI occurs when excessive fluid intake lowers the plasma osmolarity to the point that it falls below the threshold for ADH secretion.3 This condition may be associated with psychiatric disorders but also has been found in individuals who have a low osmotic threshold for inducing thirst. Chronic ingestion of extremely large quantities of fluid dilute the renal medullary concentration gradient, which results in a partial resistance to ADH. This condition resolves with effective management of water ingestion.
The signs and symptoms of DI include polyuria, nocturia, continuous thirst, and polydipsia. Untreated individuals with long-standing DI may develop a large bladder capacity and hydronephrosis (see Chapter 38). Idiopathic neurogenic DI usually has an abrupt onset, and many individuals can specifically recall the date of onset of their symptoms. Those with posttraumatic or postneurosurgical DI may develop a classic three-phase syndrome: (1) diuresis, (2) antidiuresis, and (3) polyuria/polydipsia.8 Nephrogenic DI usually has a more gradual onset.
The first step in the diagnosis of DI uses water deprivation with measurement of serum osmolarity and plasma ADH levels. Water restriction is a useful test because individuals without DI respond with a rapid decrease in urine volume and an increase in urine osmolality, whereas those with DI have no decrease in urine volume or increase in urine osmolality. In individuals with severe DI, water deprivation testing can be hazardous. If the individual loses more than 3% of the pretest body weight, circulatory collapse and shock can ensue. Neurogenic DI can be differentiated from nephrogenic DI by measuring the response to administered desmopressin. Neurogenic DI will respond with an increased ability to concentrate the urine, whereas nephrogenic DI will not respond.3 The diagnosis of dipsogenic DI can be extremely difficult, and differentiation from nephrogenic DI is based on plasma ADH levels. Copeptin, a stable fragment of the ADH precursor molecule, is a reliable surrogate measurement for ADH secretion and can be used for the diagnosis of dipsogenic DI.10
Treatment for neurogenic DI is based on the extent of the ADH deficiency and on individual variables such as age, endocrine and cardiovascular status, and lifestyle. Replacement therapy for symptomatic neurogenic DI includes administration of the synthetic vasopressin analog desmopressin acetate (DDAVP) given intranasally or orally.11
Treatment for nephrogenic DI requires treatment of any reversible underlying disorders, discontinuation of etiologic medications, and correction of associated electrolyte disorders. Although the use of thiazide diuretics has been implicated as a cause for DI, they improve salt and water absorption at the proximal tubule and may be helpful in moderate DI.3 New treatments for genetic abnormalities involving the V2 receptor are being evaluated.9,12
Diseases of the Anterior Pituitary
Disorders of the anterior pituitary may involve either hypofunction or hyperfunction of the gland.
Hypopituitarism
Hypopituitarism can be characterized by the absence of selective pituitary hormones or the complete failure of all pituitary hormone functions. Hypopituitarism results from either an inadequate supply of hypothalamic-releasing hormones, damage to the pituitary stalk, or an inability of the gland to produce hormones. The most common causes of hypopituitarism lie within the pituitary gland itself. An important cause is pituitary infarction resulting from severe shock, pituitary apoplexy, aneurysms, sickle cell disease, or pregnancy.13,14 Space-occupying lesions, such as pituitary adenomas or aneurysms, also can compress the gland. Head trauma is another important cause of panhypopituitarism and may be transient or permanent.15,16 Other causes of hypopituitarism include surgical removal or destruction of the gland, infections (e.g., meningitis, syphilis, tuberculosis), sarcoidosis, autoimmune hypophysitis, or certain drugs (e.g., bexarotene, carbamazepine).14 A rare form of hypopituitarism is characterized by combined hormonal deficiencies and is related to mutations of the prophet of pituitary transcription factor (PROP-1) gene involved in early embryonic pituitary development.17,18
Pathophysiology
The pituitary gland is highly vascular and is therefore extremely vulnerable to ischemia and infarction. It relies heavily on portal blood flow from the hypothalamus. In traumatic brain injury, disruption of blood flow can cause infarction with subsequent necrosis and fibrosis of pituitary tissue.14 After tissue necrosis, edema with swelling of the gland occurs. Expansion of the pituitary within the fixed compartment of the sella turcica further impedes blood supply to the pituitary. Over time the pituitary undergoes shrinkage, and symptoms of hypopituitarism develop.
Clinical Manifestations
ACTH deficiency is a potentially life-threatening disorder because cortisol is required for many aspects of cellular metabolism. ACTH deficiency is usually encountered with generalized pituitary hypofunction and rarely occurs as an isolated event.19 Within 2 weeks of the complete absence of ACTH, symptoms of cortisol insufficiency develop, including nausea, vomiting, anorexia, fatigue, and weakness. Hypoglycemia is caused by increased insulin sensitivity, decreased glycogen reserves, and decreased gluconeogenesis associated with hypocortisolism. In women, loss of body hair and decreased libido may be caused by decreased adrenal androgen production. ACTH deficiency also limits maximum aldosterone secretion, although the renin-angiotensin system can stimulate some aldosterone secretion with resultant hypotension and decreased urine output.
TSH deficiency also is rarely seen in isolation but most often occurs in conjunction with other pituitary hormone deficiencies. The symptoms of decreased TSH levels may become apparent in 4 to 8 weeks and may include cold intolerance, dryness of skin, mild myxedema, lethargy, and decreased metabolic rate. The symptoms are usually less severe than those associated with primary hypothyroidism, in which lack of thyroxine is related to disease in the thyroid gland (see p. 728).
GH deficiency occurs in children and adults. As described previously, GH deficiency may result from any of the causes of hypopituitarism. In children it may be genetic or it may be the result of tumors such as craniopharyngiomas. Several genetic defects have been identified in the GH axis that account for impaired GH action. These include a recessive mutation in the GHRH gene, resulting in a failure of GH secretion, and mutations that cause GH insensitivity by affecting the GH receptor, insulin-like growth factor 1 (IGF-1) biosynthesis, IGF-1 receptors, or defects in GH signal transduction.20,21 GH deficiency in children is manifested by growth failure (Figure 22-2). Another feature of GH deficiency in children is fasting hypoglycemia, likely attributable to impaired substrate mobilization for gluconeogenesis and enhanced insulin sensitivity.
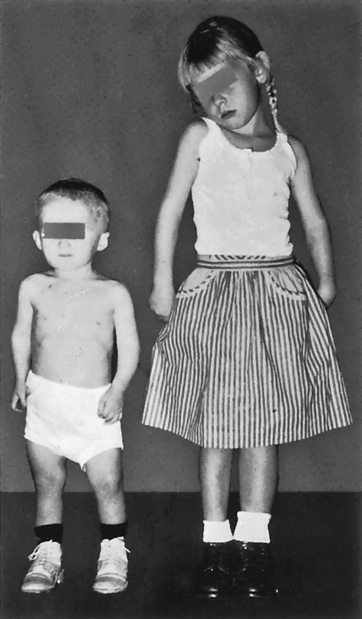
A 4-year-old boy whose height is 25 inches. The girl is also 4 years old and has a normal height of 39 inches. Dwarf has a normal face, as well as head, trunk, and limbs of approximately normal proportions. (From Brashear HR, Raney RB: Shands’ handbook of orthopaedic surgery, ed 10, St Louis, 1986, Mosby.)
In both children and adults, acute GH and IGF-1 deficiency has been implicated in significant metabolic perturbations seen with critical illness.22 Symptoms of chronic adult GH deficiency syndrome include increased body fat, decreased muscle bulk and strength, reduced sweating, dry skin, and psychologic problems, including depression, social withdrawal, fatigue, loss of motivation, and a diminished feeling of well-being.14 Several studies also have documented increased mortality in adults who are GH deficient. Osteoporosis and alterations in body composition (i.e., reduced lean body mass) are common concomitants of adult GH deficiency. A decline in GH production is an inevitable consequence of aging.23
Evaluation and Treatment
The diagnostic evaluation of suspected pituitary disease is often challenging and must be carefully interpreted together with the individual’s signs and symptoms. Simultaneous measurements of the tropic hormones from the pituitary and target endocrine glands are crucial and, in some cases, dynamic testing of the various axes is indicated.14 Radiographic assessment of the pituitary (magnetic resonance imaging [MRI] or computed tomography [CT] scans) may demonstrate enlargement of the pituitary, abnormal areas of enhancement suggestive of an adenoma, deviation of the pituitary stalk, or evidence of a locally aggressive tumor.
In general, treatment of hypopituitarism involves replacing target gland hormone(s) that are deficient. In cases of circulatory collapse, immediate therapy with glucocorticoids and intravenous fluids is critical. Thyroid and cortisol replacement therapy must be maintained. Gender-specific sex steroid replacement therapy is also initiated to improve general well-being and to prevent osteoporosis. Recombinant human GH (rhGH) is used for treatment of GH deficiency in children and results in both improved stature and metabolism.24 Treatment of adult GH deficiency improves quality of life, metabolism, body composition, and physical performance in some individuals.14,25 The use of GH to treat otherwise healthy aging is more controversial.23
Hyperpituitarism: Primary Adenoma
Pituitary adenomas are usually benign slow-growing tumors that arise from cells of the anterior pituitary, most commonly those that secrete GH and prolactin. They are very common in the population with prevalence rates estimated to be around 17%. However, most are microadenomas found incidentally on high-resolution MRI scanning and are hormonally silent and do not pose significant hazards to the individual.26,27 More significant adenomas are associated with morbidity and mortality attributable to alterations in hormone secretion or to invasion or impingement of surrounding structures. The pathogenesis of pituitary adenomas includes hypothalamic and intrapituitary factors, including altered expression of pituitary cell cycle genes, activation of pituitary selective oncoproteins, or loss of pituitary suppressor factors.28 Several associated gene mutations have been identified, including those associated with multiple endocrine neoplasia (MEN) syndromes.29 Primary pituitary carcinomas are rare, representing about 0.2% of all pituitary tumors.
Evaluation and Treatment
Diagnosis of pituitary adenoma involves physical and laboratory evaluations, including pertinent hormone assays and radiographic examination of the skull (MRI [preferred] or contrast-enhanced CT). The goal of treatment is to protect the individual from the effects of tumor growth, to control hormone hypersecretion (if present), and to replace deficient hormones while minimizing damage to appropriately secreting portions of the pituitary. Depending on the tumor size and type, individuals may be treated with specific medications to suppress tumor growth, transsphenoidal tumor resection, or radiation therapy. Simple observation and close follow-up is commonly employed for individuals who have no evidence of hormonal hypersecretion and no suggestion of anatomic aggressiveness.27,30
Hypersecretion of Growth Hormone: Acromegaly
Acromegaly occurs in adults who are exposed to continuously excessive levels of GH and concomitant elevation of IGF-1. In children and adolescents whose epiphyseal plates have not yet closed, the effect of increased GH levels on long bone growth is termed giantism (Figure 22-3).

A pituitary giant and dwarf contrasted with normal-size men. Excessive secretion of growth hormone by the anterior lobe of the pituitary gland during the early years of life produces giants of this type, whereas deficient secretion of this hormone produces well-formed dwarfs. (From Patton K, Thibodeau GA: Anatomy & physiology, ed 8, St Louis, 2013, Mosby.)
Pathophysiology
With a GH-secreting adenoma, the usual GH baseline secretion pattern and sleep-related GH peaks are lost, and an unpredictable secretory pattern ensues. With only slight elevations of GH, IGF-1 levels increase, stimulating growth. In children and adolescents whose epiphyseal plates have not yet closed, the effect of increased GH levels causes excessive skeletal growth, with some individuals becoming 8 or 9 feet tall. In the adult, epiphyseal closure has occurred and increased amounts of GH and IGF-1 cause connective tissue proliferation and increased cytoplasmic matrix, as well as bony proliferation that results in the characteristic appearance of acromegaly (Figures 22-4 and 22-5).

Chronologic sequence of photographs showing slow development of acromegaly. (From Belchetz P, Hammond P: Mosby’s color atlas and text of diabetes and endocrinology, Edinburgh, 2003, Mosby.)
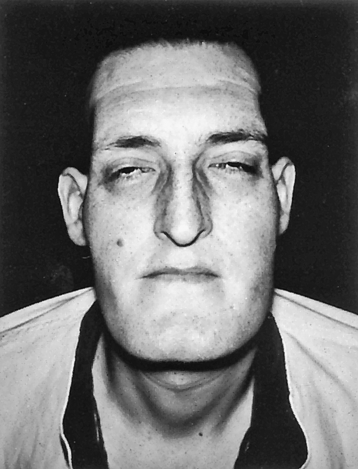
Note large head, forward projection of jaw, and protrusion of frontal bone. (Thibodeau GA, Patton K: The human body in health & disease, ed 5, St Louis, 2010, Mosby.)
GH also has significant effects on glucose, lipid, and protein metabolism.31 Hyperglycemia results from GH’s inhibition of peripheral glucose uptake and increased hepatic glucose production, followed by compensatory hyperinsulinism and, finally, insulin resistance.32 Excessive levels of GH and IGF-1 also affect the cardiovascular system. Although the associated pathophysiology is not clearly understood, hypertension and left heart failure are seen in one third to one half of individuals with acromegaly. Cardiomyopathy associated with progressive and unrestrained myocardial growth is a significant factor.33,34
Clinical Manifestations
As a result of connective tissue proliferation, individuals with acromegaly have enlarged tongues, interstitial edema, increase in the size and function of sebaceous and sweat glands (leading to increased body odor), and coarse skin and body hair. The coarse skin condition becomes very apparent when procedures such as inserting an intravenous needle are performed; the skin is very thick and difficult to penetrate. Bony proliferation results in large joint arthropathy with swelling and decreased range of motion and periosteal vertebral growth, which causes kyphosis. Enlargement of the facial bones and the bones of the hands and feet result in protrusion of the lower jaw and forehead and a need for increasingly larger sizes of shoes, hats, rings, and gloves (see Figures 22-4 and 22-5).
Evaluation and Treatment
Diagnosis of acromegaly is accomplished by documenting elevated IGF-1 levels and GH suppression during oral glucose tolerance testing. The goals of treatment are to normalize GH and IGF-1 serum levels, restoring normal pituitary function and relieving or preventing complications related to tumor expansion. The treatment of choice for acromegaly is transsphenoidal surgery or endonasal endoscopic surgery for removal of the GH-secreting adenoma.35 Treatment by radiation therapy may be effective when rapid control of GH levels is not essential, when the individual is not a good surgical candidate, or when hyperfunction persists after subtotal resection. Octreotide, octreotide long-acting, and lanreotide are somatostatin analogs that have been shown to be effective in lowering elevated GH levels, reversing many of the clinical manifestations of the disease, and causing tumor shrinkage.36 Dopaminergic agonists, such as cabergoline, also may be helpful, especially if the tumor also secretes prolactin. Pegvisomant is an effective drug that induces tissue insensitivity to GH by blocking the GH receptor.37
Hypersecretion of Prolactin: Prolactinoma
Pituitary tumors that secrete prolactin are called prolactinomas and are the most common of the hormonally active pituitary tumors encountered in clinical medicine. These tumors can be classified as microprolactinomas (<1 cm in size) or macroprolactinomas (>1 cm in size). Microprolactinomas are usually encapsulated and noninvasive, whereas macroprolactinomas commonly expand into the optic chasm and invade local structures.38 The physiologic actions of prolactin include breast development during pregnancy, postpartum milk production, and suppression of ovarian function in nursing women.
In addition to pituitary tumors, many conditions or medications can elevate prolactin level in the absence of pituitary pathologic condition. For example, renal failure, hepatic cirrhosis, polycystic ovarian disease (see Chapter 24), breast stimulation, or chest wall lesions can increase prolactin levels. Because thyrotropin-releasing hormone (TRH) stimulates prolactin secretion, in addition to enhancing TSH release, prolactin level may be elevated in individuals with primary hypothyroidism. Prolactin is under tonic inhibitory hypothalamic control through the secretion of dopamine (prolactin inhibitor factor [PIF]). Thus medications that block the effects of dopamine at the pituitary can increase prolactin production and stimulate proliferation of prolactin-secreting cells (lactotropes). These include antipsychotics (risperidone, chlorpromazine), metoclopramide, tricyclic antidepressants, methyldopa, and estrogens. Any process that interferes with the delivery of dopamine from the hypothalamus to the lactotropes (pituitary stalk tumor, pituitary stalk transection, or compressive pituitary tumor) also results in hyperprolactinemia.
Clinical Manifestations
Women with hyperprolactinemia generally present with galactorrhea (nonpuerperal milk production) and menstrual disturbances, including amenorrhea. In susceptible women, hirsutism develops because of estrogen deficiency. If not detected until after many years, this estrogen deficiency also may result in osteoporosis. Men often develop hypogonadism and erectile dysfunction but may not seek care until symptoms related to the increasing size of the adenoma (i.e., headache or visual impairment) develop.39
Evaluation and Treatment
Dopaminergic agonists (bromocriptine, cabergoline, and pergolide) are the treatment of choice for prolactinomas, and their use is often associated with both a rapid reduction in the size of the tumor and a reversal of the gonadal effects of hyperprolactinemia.40 Restoration of fertility in previously anovulatory women is common. Although there is an association between valvular heart disease and cabergoline used in the treatment of parkinsonism, a similar association has not been found when cabergoline is used to treat prolactinoma.41,42 In individuals resistant or intolerant to these medications, transsphenoidal surgery, endonasal endoscopic surgery, and radiotherapy are options.38
Alterations of Thyroid Function
result of pituitary or hypothalamic alterations. Primary thyroid disorders result in alterations of thyroid hormone (TH) levels with secondary feedback effects on pituitary TSH levels. For example, when there are primary elevations in TH level, TSH level will secondarily decrease because of negative feedback. When TH level is decreased because of a condition affecting the thyroid gland, TSH level will be elevated. Thyroid disease also can present with no or minimal symptoms but with abnormal laboratory values. This is known as subclinical thyroid disease. Central (secondary) thyroid disorders are related to disorders of pituitary gland TSH production. When there is excessive TSH production, TH level is elevated secondary to the primary elevation of TSH level. The reverse is true with inadequate TSH production. Exposure to iodinated contrast media has been shown to be associated with development of both hyperthyroidism and hypothyroidism (see What’s New? Thyroid Disease and Iodinated Contrast Media [ICM]).
Hyperthyroidism
Thyrotoxicosis
Pathophysiology
Thyrotoxicosis is a condition that results from any cause of increased amounts of TH. Hyperthyroidism is a form of thyrotoxicosis in which excess amounts of TH are secreted from the thyroid gland. The terms thyrotoxicosis and hyperthyroidism are often used interchangeably. The prevalence of hyperthyroidism is estimated to be 0.7% to 2.1% in the United States, of which 0.7% is subclinical and is more prevalent in women and in iodine-deficient geographical areas.43 Common diseases that cause primary hyperthyroidism include Graves disease, toxic multinodular goiter, a solitary toxic adenoma (Figure 22-6), and, very rarely, follicular thyroid carcinoma. Central (secondary) hyperthyroidism is less common and is caused by TSH-secreting pituitary adenomas. Thyrotoxicosis not associated with hyperthyroidism includes subacute thyroiditis, ectopic thyroid tissue, and ingestion of excessive TH. TSH progressively decreases in subclinical hyperthyroidism and can be caused by thyroid hormone therapy or iatrogenic exogenous disease. It is more prevalent in iodine-deficient populations.44 Identifying the cause is important because the treatment and expected outcome vary accordingly. Each condition is associated with specific pathophysiology and manifestations; however, all forms of thyrotoxicosis share some common characteristics.
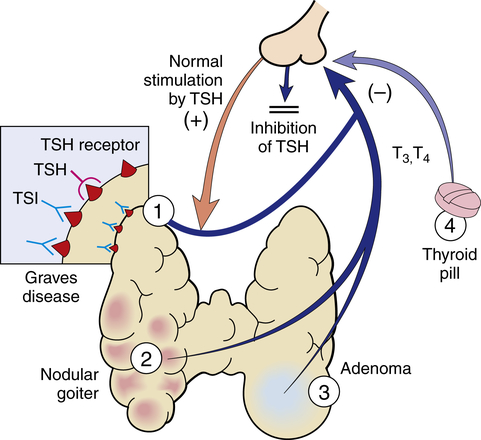
Hyperthyroidism may have several causes, among them: 1, Graves disease; 2, toxic multinodular goiter; 3, follicular adenoma; 4, thyroid medication. TSH, Thyroid-stimulating hormone; TSI, thyroid-stimulating immunoglobulin. (From Damjanov I: Pathology for the health professions, ed 4, St Louis, 2012, Saunders.)
Clinical Manifestations
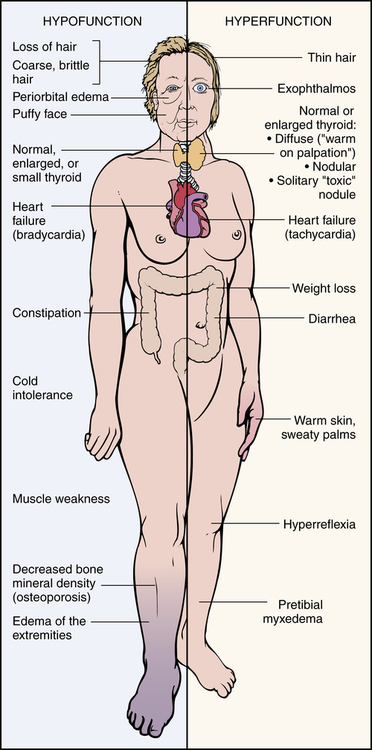
The clinical features of thyrotoxicosis are attributable to the metabolic effects of increased circulating levels of TH (Figure 22-7). This usually results in an increased metabolic rate with heat intolerance and increased tissue sensitivity to stimulation by the sympathetic division of the autonomic nervous system. The major manifestations are summarized in Table 22-2. Enlargement of the thyroid gland (goiter) is common in hyperthyroid conditions caused by stimulation of TSH receptors.
TABLE 22-2
SYSTEMIC MANIFESTATIONS OF HYPERTHYROIDISM
| SYSTEM | CLINICAL MANIFESTATIONS | MECHANISMS UNDERLYING CLINICAL MANIFESTATIONS |
| Endocrine | Enlarged thyroid gland (goiter) (97-99% of cases); systolic or continuous bruit over thyroid; increased cortisol degradation; hypercalcemia and decreased PTH secretion; diminished sensitivity to exogenous insulin | Hyperactivity of the thyroid gland; excess bone resorption leading to hypercalcemia and a disruption of PTH-regulating mechanisms; increased insulin degradation |
| Reproductive | Oligomenorrhea or amenorrhea; erectile dysfunction and decreased libido; increased serum estradiol and estrone levels but lower than normal levels of free estradiol and estrone | Menstrual cycle alterations that may be related to hypothalamic or pituitary disturbances; increase in sex hormone–binding globulin |
| Gastrointestinal | Weight loss; increased peristalsis leading to less formed and more frequent stools; nausea, vomiting, anorexia, abdominal pain; increased use of hepatic glycogen stores and of adipose and protein stores; decrease in serum lipid levels (including triglycerides, phospholipids, and cholesterol); changes in vitamin metabolism leading to decrease in tissue stores of vitamins | Increased catabolism leading to the body’s inability to meet its metabolic needs; increased glucose absorption; increase in cholesterol excretion in feces and cholesterol conversion to bile salts; impaired conversion of B vitamins to their coenzymes, causing increased need for water-soluble and fat-soluble vitamins |
| Integumentary | Excessive sweating, flushing, and warm skin; heat intolerance; hair fine, soft, and straight; temporary hair loss; nails that grow away from nail beds, palmar erythema | Hyperdynamic circulatory state |
| Sensory (eyes) | Ocular manifestations including elevated upper eyelid leading to decreased blinking and a staring quality; fine tremor of lid; infiltrative ocular changes associated with Graves disease | Overactivity of Müller muscle; inflammation of retroorbital contents |
| Cardiovascular | Increased cardiac output and decreased peripheral resistance; tachycardia at rest; loud heart sounds; supraventricular dysrhythmias, left ventricular dilation and hypertrophy | Hypermetabolism and need to dissipate heat |
| Nervous | Restlessness; short attention span; compulsive movement; fatigue; tremor; insomnia; increased appetite; emotional lability | Not clearly defined; alterations in cerebral metabolism resulting from excess thyroid hormone |
| Pulmonary | Dyspnea; reduced vital capacity | Weakness of respiratory muscles |
Evaluation and Treatment
The diagnosis of thyrotoxicosis is based on symptoms of TH excess and documentation of increased circulating thyroid hormone levels. Elevated serum thyroxine (T4) and triiodothyronine (T3) levels and decreased serum TSH levels are diagnostic for primary hyperthyroidism. By contrast, central (secondary) hyperthyroidism caused by TSH-secreting pituitary tumors is characterized by normal to increased TSH levels in the face of elevated thyroid hormone concentrations. Radioactive iodine is used to test for increased uptake in primary hyperthyroidism (Figure 22-8).
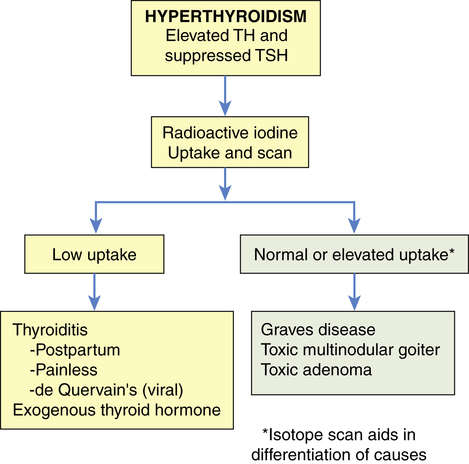
Radioactive iodine is used in the differential diagnosis of hyperthyroidism. TH, Thyroid hormone; TSH, thyroid-stimulating hormone.
Treatment is directed at controlling excessive TH production, secretion, or action. The major types of therapy currently used to achieve these goals include antithyroid drug therapy (methimazole or propylthiouracil), radioactive iodine therapy, and surgery. Guidelines for the treatment of hyperthyroidism and thyrotoxicosis have been published.45 Treatment of subclinical hyperthyroidism is based on levels of TSH and clinical symptoms.44 A major complication of all forms of treatment for hyperthyroidism is hypothyroidism.46
Hyperthyroid Conditions
Graves Disease
Graves disease is the underlying cause of 50% to 80% of cases of hyperthyroidism and has a prevalence of approximately 0.5% in the U.S. population.47 It occurs more commonly in women. Although the cause of Graves disease is not known, genetic factors interacting with environmental triggers play an important role in the pathogenesis of this autoimmune thyroid disease. Graves disease results from a form of type II hypersensitivity (see Chapter 9) in which there is stimulation of the thyroid by autoantibodies directed against the TSH receptor. These autoantibodies, called thyroid-stimulating immunoglobulins (TSIs; also called thyroid-stimulating antibodies [TSAbs] or thyroid receptor antibodies [TRAbs]), override normal regulatory mechanisms. The TSI stimulation of TSH receptors in the gland results in hyperplasia of the gland (goiter) and increased synthesis of TH, especially of triiodothyronine (T3). Increased levels of TH affect every physiologic system and result in the classic signs and symptoms of hyperthyroidism illustrated in Figure 22-8. TSH production by the pituitary is inhibited through the usual negative feedback loop.
TSIs also contribute to two major distinguishing clinical manifestations of Graves disease: ophthalmopathy and pretibial myxedema (Graves dermopathy). Graves ophthalmopathy affects more than half of individuals with Graves disease. There are two categories of ocular manifestations: (1) functional abnormalities resulting from hyperactivity of the sympathetic division of the autonomic nervous system (lag of the globe on upward gaze or a lag of the upper lid on downward gaze) and (2) infiltrative changes involving the orbital contents with enlargement of the ocular muscles. The infiltrative changes result from TSH receptor autoantibodies reacting with receptors on orbital fibroblasts (Figure 22-9). Increased secretion of hyaluronic acid, orbital fat accumulation, inflammation, and edema of the orbital contents result in exophthalmos (protrusion of the eyeball). Periorbital edema and extraocular muscle weakness lead to diplopia (double vision). The individual may experience irritation, pain, lacrimation, photophobia, blurred vision, decreased visual acuity, papilledema, visual field impairment, exposure keratosis, and corneal ulceration.48
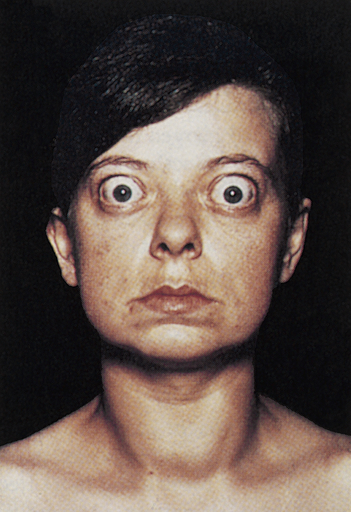
Note large and protruding eyeballs in association with a large goiter. (From Stein HA, Slatt BJ, Stein RM: The ophthalamic assistant: fundamentals and clinical practice, ed 7, Philadelphia, 2000, Mosby.)
A small number of individuals with Graves disease and very high levels of TSI experience pretibial myxedema (Graves dermopathy), characterized by subcutaneous swelling on the anterior portions of the legs and by indurated and erythematous skin (Figure 22-10). Graves dermopathy is associated with thyrotropin receptor antigens on fibroblasts and recruited T lymphocytes that stimulate excess amounts of hyaluronic acid production in the dermis and subcutis. The manifestations occasionally appear on the hands giving the appearance of clubbing of the fingers (thyroid acropachy).49
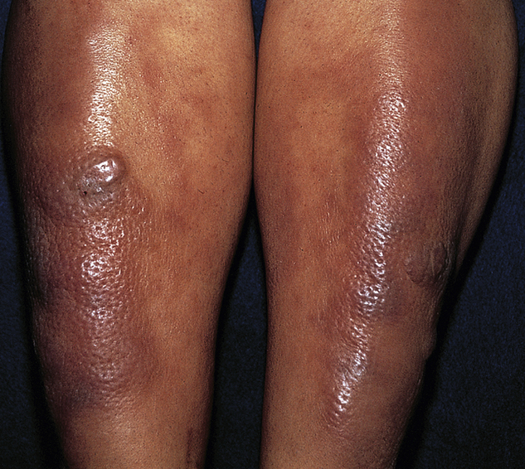
Purple-brown edematous plaques on shins of individual with Graves disease. Note orange peel appearance. (From Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology, ed 3, New York, 2013, Saunders.)
Therapy for Graves disease includes antithyroid drugs, radioactive iodine (used with caution in Graves ophthalmopathy because it may worsen the condition), or surgery. Unfortunately, current treatment for Graves disease does not reverse the infiltrative ophthalmopathy or the pretibial myxedema. Surgical orbital decompression and glucocorticoids can help many individuals with progressive ophthalmopathy.50 Skin lesions rarely require treatment, but if they are symptomatic they may respond to topical glucocorticoids.51
Hyperthyroidism Resulting from Nodular Thyroid Disease
Irreversible changes may occur in some follicular cells and these cells function autonomously and produce excessive amounts of TH. On the other hand, some follicular cells may cease to function. The balance between the amount of TH produced by hyperfunctioning nodules and that produced by the remainder of the gland determines whether an individual becomes euthyroid or hyperthyroid. Toxic multinodular goiter occurs when there are several hyperfunctioning nodules leading to hyperthyroidism. If only one nodule becomes hyperfunctioning it is termed solitary toxic adenoma. This condition is more common in iodine-deficient regions. The classic clinical manifestations of hyperthyroidism (see Figure 22-7) usually develop slowly and exophthalmos and pretibial myxedema do not occur. Nodules may be palpable on physical examination. The incidence of malignancy in toxic multinodular goiter is estimated to be as high as 9% and 10.5%, so most individuals should undergo a fine needle aspiration biopsy of suspicious nodules before treatment.52 Treatment consists of a combination of radioactive iodine, surgery, or antithyroid drugs. Mutations of the TSH receptor have been found in most solitary toxic adenomas.53,54
Thyrotoxic Crisis (Thyroid Storm)
Thyrotoxic crisis (thyroid storm) is a rare but dangerous worsening of the thyrotoxic state, in which death can occur within 48 hours without treatment. The condition may develop spontaneously, but it occurs in individuals who have undiagnosed or partially treated severe hyperthyroidism and who are subjected to excessive stress from other causes. These causes may include infection, pulmonary or cardiovascular disorders, trauma, burns, seizures, surgery (especially thyroid surgery), obstetric complications, emotional distress, or dialysis. The symptoms of thyroid crisis are caused by the increased action of thyroxine (T4) and triiodothyronine (T3) exceeding metabolic demands.55
Hypothyroidism
Hypothyroidism is caused by a deficient production of TH by the thyroid gland. Hypothyroidism is the most common disorder of thyroid function; it affects between 0.1% and 2% of individuals in the United States and is more common in women and the elderly. It may be primary or secondary. Primary hypothyroidism is the most prevalent. Central (secondary) hypothyroidism, which is much less common, includes conditions that cause either pituitary or hypothalamic failure with failure to stimulate normal thyroid function. Subclinical hypothyroidism is mild thyroid failure and is estimated to occur in 4% to 8% of U.S. adults. It is defined as an elevation in TSH level with normal levels of circulating TH.44
Pathophysiology
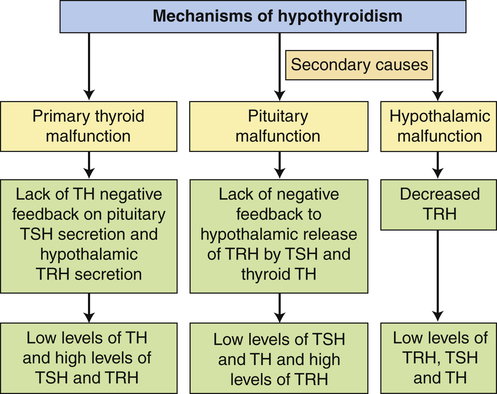
In primary hypothyroidism the loss of functional thyroid tissue leads to a decreased production of TH. Causes in adults include autoimmune thyroiditis (Hashimoto disease), iatrogenic loss of thyroid tissue after surgical or radioactive treatment for hyperthyroidism, head and neck radiation therapy, medications, and endemic iodine deficiency. Infants and children may present with hypothyroidism because of congenital defects in the pituitary or thyroid glands.56 Most states require newborn screening for hypothyroidism. Central (secondary) hypothyroidism is caused by the pituitary’s failure to synthesize adequate amounts of TSH or a lack of TRH. Pituitary tumors that compress surrounding pituitary cells or the consequences of their treatment are the most common causes of secondary hypothyroidism. Other causes include traumatic brain injury, subarachnoid hemorrhage, or pituitary infarction. Hypothalamic dysfunction results in low levels of TRH, TSH, and TH (Figure 22-11).
Clinical Manifestations
Hypothyroidism generally affects all body systems and occurs insidiously over months or years. The extent of the symptoms is closely related to the degree of TH deficiency (see Figure 22-7). The lowered levels of TH result in decreased energy metabolism and decreased heat production. The individual develops a low basal metabolic rate, cold intolerance, lethargy, tiredness, and slightly lowered basal body temperature and also may have diastolic hypertension. Many organ systems are affected (Table 22-3). The decrease in the level of TH leads to increases in TSH production and may cause goiter.
TABLE 22-3
SYSTEMIC MANIFESTATIONS OF HYPOTHYROIDISM
| SYSTEM | CLINICAL MANIFESTATIONS | MECHANISMS UNDERLYING CLINICAL MANIFESTATIONS |
| Neurologic | Confusion, syncope, slowed speech and thinking, memory loss; lethargy, headaches, hearing loss, night blindness; slow, clumsy movements; cerebellar ataxia; slow alpha-wave activity and loss of amplitude in EEG; reduced cAMP response to epinephrine, glucagons, and PTH stimulation; decreased appetite | Decreased cerebral blood flow leading to cerebral hypoxia; reduced intracellular processes caused by decreased β-adrenergic activity that may be related to a decrease in the number of β-adrenergic receptor sites |
| Endocrine | Increased TSH production in primary hypothyroidism; enlarged pituitary thyrotropes, increase in serum prolactin levels with galactorrhea; decreased rate of cortisol turnover but with normal serum cortisol levels | Impaired TH synthesis or defects in iodide trapping leading to compensatory TSH production; chronic overstimulation of thyrotropes of TRH and by TSH synthesis; stimulation of lactotropes by TRH related to increased prolactin levels; decreased deactivation of cortisol |
| Reproductive | Decreased androgen secretion in men, increased estriol formation in women; low total hormone values but with increased amounts of unbound hormone; anovulation, decreased libido, menorrhagia, and a high incidence of spontaneous abortion in women; erectile dysfunction, decreased libido, and oligospermia in men | Altered metabolism of estrogens and androgens; decreased levels of sex hormone–binding globulin |
| Hematologic | Decrease in red cell mass leading to normocytic, normochromic anemia; macrocytic anemia associated with vitamin B12 deficiency and inadequate folate or iron absorption in the gastrointestinal tract | Decreased basal metabolic rate and reduced oxygen requirements; decreased production of erythropoietin; possible relationship between TH and optimal hematologic response to vitamin B12 |
| Cardiovascular | Reduction in stroke volume and heart rate causing lowered cardiac output; increased peripheral vascular resistance to maintain systolic blood pressure can cause hypertension; normal response to exercise but with alterations in circulatory system at rest (prolonged circulation time and decreased blood flow to tissues); cool skin and cold tolerance; enlarged heart; decreased intensity of heart sounds and variety of ECG changes (sinus bradycardia, prolonged PR interval, depressed P waves, flattened or inverted T waves, and low-amplitude QRS complexes); cardiac tamponade (although rare) (see Chapter 32) | Decreased metabolic demands and loss of regulatory and rate-setting effects of TH; protein-mucopolysaccharide–rich fluid in the pericardial sac associated with enlarged heart; pericardial effusions associated with heart sounds and ECG changes Increases in peripheral vascular resistance and increased blood volume can cause hypertension |
| Pulmonary | Dyspnea; myxedematous changes in respiratory muscles leading to hypoventilation and carbon dioxide retention, which contribute to myxedema coma | Pleural effusions associated with dyspnea, although effusions may be asymptomatic |
| Renal | Reduced renal blood flow and glomerular filtration rate leading to decreased renal excretion of water; increase in total body water and dilutional hyponatremia; reduced production of erythropoietin | Hemodynamic alterations associated with reduced blood flow and filtration; increased total body water related to decreased excretion and mucinous deposits in tissue |
| Gastrointestinal | Constipation, weight gain, and fluid retention; decreased absorption of most nutrients; decreased protein metabolism leading to retarded skeletal and soft tissue growth and slightly positive nitrogen balance; edema; decreased glucose absorption and delayed glucose uptake; elevated serum lipid values | Reduced intake and reduced peristaltic activity that may progress to fecal impaction; water absorption related to prolonged transit time; fluid retention associated with myxedematous changes; edema associated with high concentrations of exchangeable albumin in the extravascular space caused by increased capillary permeability to proteins; depressed insulin degradation; depressed lipid synthesis and degradation |
| Musculoskeletal | Muscle aching and stiffness; slow movement and slow tendon jerk reflexes; decreased bone formation and resorption, increased bone density; aching and stiffness in joints | Decreased rate of muscle contraction and relaxation contributing to slow movement and reflexes |
| Integumentary | Dry, flaky skin; dry, brittle head and body hair; reduced growth of nails and hair; slow wound healing | Reduced sweat and sebaceous gland secretion |
| Myxedema | Accumulation of hyaluronic acid, which binds water and causes a puffy appearance | |
| Cool skin | Decreased circulation to skin |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


