Figure 12.1
FGFR-associated syndromic conditions
Table 12.1
FGFR1, –2, and –3 genes
Gene | OMIM | Chromosomal location | Gene organization |
|---|---|---|---|
FGFR1 | *136350 | 8p11.23-p11.22 | 18 exons, 17 coding |
FGFR2 | *176943 | 10q26.13 | 18 exons, 17 coding |
FGFR3 | *134934 | 4p16.3 | 18 exons, 17 coding |
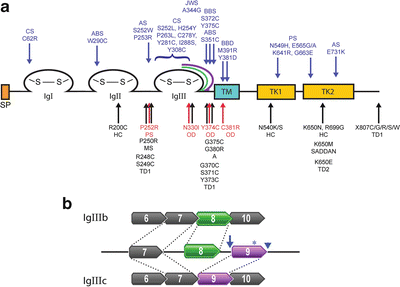
Figure 12.2
FGFR protein structure. (a) Common FGFR protein structure showing the signal peptide domain (SP, orange), the three immunoglobulin loops (IgI, IgII, and IgIII) with cysteine–cysteine disulfide bonds (S–S), the transmembrane domain (TM, blue), and the bifurcated tyrosine kinase domain (TK1 and TK2, yellow). The alternatively spliced carboxy-region of IgIII is illustrated as green and purple lines. Locations of selected mutations in FGFR1 (red), FGFR2 (blue), and FGFR3 (black) are indicated by arrows, with the associated disease above or below the mutation. A achondrodysplasia, ABS Antley-Bixler syndrome, AS Apert syndrome, BBD bent bone dysplasia, BSS Beare-Stevenson syndrome, CS Crouzon syndrome, HC hypochondrodysplasia, JWS Jackson-Weiss syndrome, MS Muenke syndrome, OD osteoglophonic dysplasia, PS Pfeiffer syndrome, TD1 and TD2 thanatophoric dysplasia type 1 and 2, respectively. (b) Alternative splicing of exons 8 and 9 that code for the IgIII carboxy-terminal region. Isoform FGFR-IIIb (top) includes exon 8 (green). Isoform FGFR-IIIc (bottom) includes exon 9 (purple). Examples of FGFR2 splicing mutations are shown: c.940-2A>G (arrow), c.1032G>A (A344A) (asterisk); and c.1084+3A>G (arrowhead)
Molecular Basis of Disease
Mutations in the FGFR1, –2, and –3 genes account for approximately 15–20 % of all craniosynostosis and chondrodysplasias. FGFR-related skeletal anomalies are a result of gain-of-function variants that constitutively activate the receptor function [16–18]. Activated FGFRs receptors cause increased cellular proliferation and premature osteoblast differentiation [19–21]. The FGFR constitutive activation is by either a FGF ligand-dependent or -independent mechanism. Ligand-dependent mechanisms arise due to mutations that improve binding of FGF ligands and dimerization of the receptors prolonging signaling activity [22]. Alternatively, ligand-independent mechanisms include the following: (1) enhancement of receptor dimerization due to an immunoglobulin domain structural change such as a gain or loss of a cysteine residue within the loops; (2) augmentation of dimerization due to intramembrane domain changes in amino acid charge that increases hydrogen bonding; and (3) alterations in the kinase domain causing constitutively active phosphorylation [23–25].
Mutations causing FGFR-related craniosynostosis and/or chondrodysplasia tend to cluster in specific domains of the receptors (Fig. 12.2a). These clusters illustrate the importance of the domains in receptor function. Analogous mutations found in each of the different FGFRs tend to mirror phenotypic effects between the receptors. However, distinctions between analogous mutations in the different receptors and their phenotypes reveal their independent roles during normal skeletogenesis.
Craniosynostosis and Chondrodysplasia
Craniosynostosis
Craniosynostosis arises from the premature fusion of one or more of the cranial sutures resulting in a dysmorphic skull. Distortions of the skull arise from uneven growth patterns between the sutures and depend on the location and timing of the fusion events [26]. The cranial sutures are the leading edges of the growing intramembranous bones that comprise the skull vault or calvaria. The calvaria consists of the left and right frontal, squamous occipital, squamous temporal, and the left and right parietal bones that will form the neurocranium (Fig. 12.3). At birth, the frontal bones are separated by the metopic suture. The coronal suture separates the frontal bones from the left and right parietal bones. The lambdoid suture separates the parietals from the occipital bone. The suture leading edges function as ossification centers. The calvaria of the neurocranium that forms the desmocranium ossifies as intramembranous bone forming the vault of the neurocranium. Normally, the metopic suture begins to close after the first year and closure is completed by the seventh year, generating the frontal bone [27]. The sagittal, coronal, and lambdoidal sutures generally complete fusion between 20 and 40 years of age. De novo or autosomal dominantly inherited mutations in the FGFR1–3 genes that enhance signaling result in the inhibition of the osteogenic proliferation program, thereby causing premature fusion of cranial sutures [28]. Mutations associated with isolated nonsyndromic forms of craniosynostosis have been found in each of the FGFR1–3 genes [29–31].
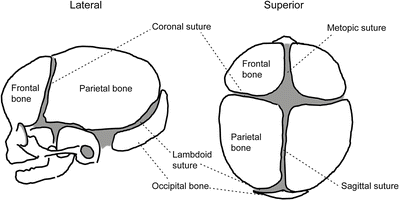
Figure 12.3
Skull sutures of a newborn. (left) Lateral view. (right) Superior view
Nonsyndromic or isolated single-suture forms of craniosynostosis are more common than syndromic and number 1 in 2,100–3,000 live births [17]. Sagittal synostosis is the most common, followed by coronal, metopic, and then lambdoid suture synostosis (Fig. 12.4) [32, 33]. Nonsyndromic craniosynostosis is heterogeneous and many of the causes remain unknown. Often full sequencing of the FGFR genes may be requested to rule out the receptors and potentially detect de novo mutations.
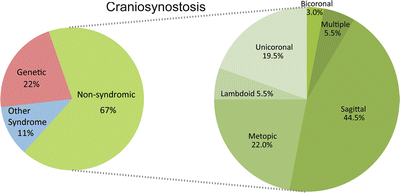
Figure 12.4
The prevalence of different forms of craniosynostosis derived from analysis of a 215-patient cohort affected with syndromic and nonsyndromic forms of craniosynostosis (left). Phenotypic categorization of nonsyndromic cases based upon affected sutures (right) (adapted with permission from Wilkie et al. 2010 [32])
Chondrodysplasias
In addition to influencing the growth of the neurocranium, FGF signaling also participates in the development of the skeletal system exemplified by its role in the growth of the long bones of the appendicular skeleton. Dominantly inherited and de novo gain-of-function mutations result in varying degrees of dwarfism as illustrated by particular FGFR3 mutations that cause hypochondroplasia and achondroplasia. Observed in both craniosynostosis and chondrodysplasia cases, germline mutations due to advanced paternal age are a significant contributor to these disorders [34, 35].
Clinical Utility of Testing
Syndromic forms of FGFR-related craniosynostosis and chondrodysplasia are diagnosed clinically, based on their well-described phenotypes. Three-dimensional ultrasonography is able to detect early skeletal anomalies, including short bones, premature skull fusion, and other characteristic syndromic features [6, 36]. Molecular testing is performed to confirm the diagnosis and provide recurrence risks in pregnancies with either suspected germline mosaic transmission in families with a history of a previously affected fetus, or in families with an affected parent and a 50 % probability of passing on the deleterious allele. In such cases of inheritance, examination of the contributing parent may reveal a mild phenotype. For severe cases, de novo mutations occurring in the male germline may be responsible. Advanced paternal age is known to increase the risk of having affected offspring. Generally, targeted mutation analysis is sufficient to confirm a well-characterized phenotype and is the first tier of testing. Sequencing of all the exons may be performed if no mutation is found initially.
Available Assays
Different methods have been developed to identify mutations within the FGFR genes. The vast majority of mutations are missense and nonsense, with splicing and in-frame small insertions and deletions being much rarer. Detection methods include targeted mutation analysis, scanning of specific exons, and sequencing of all the coding regions as well as their intron/exon boundaries. Targeted analysis is based on testing the patient’s DNA for previously described mutations. Using restriction fragment length polymorphism (RFLP) methods, regions of interest within the coding sequences can be amplified by polymerase chain reaction (PCR) and digested with restriction endonucleases that would characteristically identify specific mutations based on altered banding patterns observed by gel electrophoresis and staining (Fig. 12.5). Incomplete digestion of a restriction site may lead to aberrant results and an incorrect heterozygosity interpretation; therefore, both positive and negative digestion controls are essential for RFLP analysis. Alternatively, allele-specific oligonucleotide PCR with primers targeted to wild-type alleles and previously characterized mutations may be performed, resulting in identifiable amplicon patterns. The drawback to targeted analysis is that mutations outside the scope of the assay will be missed.
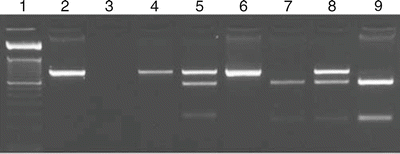
Figure 12.5
Thanatophoric dysplasia type 2 restriction fragment length polymorphism (RFLP) assay. Amplification of the FGFR3 exon 15 and digestion of patient and control DNA give characteristic banding patterns diagnostic for the p.Lys650Glu mutation. Lane 1, 50 base pair (bp) size marker; lane 2, undigested 450 bp amplicon; lane 3, no DNA template PCR control; lanes 4–6, restriction digest with BsmAI; lanes 7–9, digest with BbsI. Lanes 4, 6, 7, and 9, control DNA. Lanes 5 and 8, patient DNA. Lanes 4–6 show a gain of a BsmAI restriction site in the patient DNA (lane 5). Lanes 7–9 demonstrate loss of a BbsI digest site in the patient’s DNA (lane 8)
Mutation scanning by denaturing high-performance liquid chromatography (DHPLC) has been employed to screen the exons of the FGFR genes whereby patient’s profiles suggest potential mutations in comparison to unaffected and affected controls. Issues arise in scanning techniques such as DHPLC as well as high-temperature melting profiles and single-stranded conformational polymorphism in their inability to identify specific mutations or distinguish between known mutations and rare polymorphisms. Unique profiles identifying potential mutations must then be directly sequenced. Sequence analysis of the coding exons is the most comprehensive of the methods ensuring that any point mutation and small deletions and duplications will be identified within the amplified regions. Massively parallel sequencing, using next-generation sequencing platforms, also offers the option to perform sequence analysis on many samples concurrently through barcoding of individual patient’s DNA and using bioinformatics to separate the reads by patient and analyze the sequence data.
Deletion/duplication analysis may be performed by multiplex ligation-dependent probe amplification or exon level array-based comparative genomic hybridization and then confirmed by quantitative PCR. These methods measure the relative amounts of each exonic region where probes are placed, comparing the quantitative results to internal controls, usually housekeeping gene(s) found on other chromosomes.
Interpretation of Test Results
The majority of FGFR mutations causing skeletal dysplasia are point mutations. Interpretation of novel missense variants is becoming easier as the knowledge base grows. To date, there are approximately 155 known mutations within the FGFR1-2-3 that cause skeletal dysplasias. Current lists of mutations are available in the Human Gene Mutation Database (www.biobase-international.com/product/hgmd). Databases that include polymorphisms, such as dbSNP (www.ncbi.nlm.nih.gov/projects/SNP/), the 1000 Genomes Project (www.1000genomes.org/), and exome sequencing projects (Exome Aggregation Consortium [ExAC], exac.broadinstitute.org; Exome Variant Server, http://evs.gs.washington.edu/EVS/), provide information on the genetic variants and their frequencies in the general population [37, 38]. Variants of unknown significance can be evaluated using predictive algorithms that may provide insight into whether they are benign or pathogenic. These predictions take into consideration the biochemical properties of the amino acid change, including size, charge, polarity, steric constraints, and evolutionary conservation of the residue within the protein domain. Internet-based predictive tools, including PolyPhen2, SIFT, and Mutation Taster, provide analysis of the probable functional consequences of missense variants [39–41]. The results of these prediction tools have to be considered as one piece of evidence.
Rare splicing mutations causing skeletal dysplasia have been documented. Mutations, such as the synonymous change p.Ala344Ala (c.1032G>A) in the immunoglobulin IIIc (IgIIIc) domain of FGFR2, activate a cryptic splice donor site (Fig. 12.2b) [42]. Conversely, weakening the endogenous donor site (c.1084+3A>G and c.1084+1G>A) induces exon skipping and preferential splicing of the IgIIIb isoform [43–45]. Similarly, FGFR2 mutations causing Pfeiffer syndrome affect the IgIIIc acceptor site (c.940-2A>G or A>T) [46]. Unknown variants found in the intronic regions of the gene can be analyzed through splicing algorithms, such as the Human Splice Finder (www.umd.be/HSF/) [47] and Netgene2 (www.cbs.dtu.dk/services/NetGene2/) [48], to predict their influence on the precursor mRNA.
Laboratory Issues
Commercial test kits for analyzing the FGFR genes are not available, so testing is performed using laboratory developed tests. Test development and validation may be achieved using cell lines obtained from the Coriell Cell Repository (http://ccr.coriell.org). Proficiency testing is available through the College of American Pathologists Molecular Genetics Laboratory sequencing surveys. Additionally, interlaboratory sample exchanges and internal repeat testing of blinded samples can be used to meet the proficiency testing requirements.
FGFR-Related Skeletal Syndromes
Apert Syndrome
Apert syndrome (OMIM#101200), also known as acrocephalosyndactyly, is characterized by craniosynostosis, facial hypoplasia, broad thumbs and great toes, and digit fusion of the hands and feet (syndactyly) described as a “mitten-hand” malformation. As the most common of the craniosynostosis syndromes, Apert syndrome accounts for approximately 4.5 % of all cases with an occurrence estimated as 1 in 65,000 to 80,000 [49, 50]. The majority of cases result from de novo mutations attributed to increasing paternal age [51].
The majority of cases (99 %) are caused by one of two point mutations in the FGFR2 gene [52]. The common mutations are p.Ser252Trp, which accounts for approximately two-thirds of the cases, and p.Pro253Arg accounting for one third of cases. These mutations, located at the IgII-IgIII linker region, enhance ligand binding affinity [9]. Rarer mutations have been observed surrounding these two mutations in the linker region. Diagnostic testing can be performed by targeted Sanger sequencing.
Apert syndrome is clinically diagnosed. Identification of the specific mutation helps in determining the potential risk for further affected pregnancies due to germline mosaicism. With an affected parent, there is a 50 % probability that a pregnancy will result in an affected child.
Pfeiffer Syndrome
Patients presenting with Pfeiffer Syndrome (OMIM#101600) exhibit brachycephaly, hypertelorism, ocular proptosis, a flat midface, broad thumbs, and medially deviated great toes, and occasionally with hearing loss and variable cutaneous syndactyly [17]. Pfeiffer syndrome is a genetically heterogeneous disorder. Mutations have been found in both FGFR1 and FGFR2. FGFR2 mutations account for 95 % of patients’ mutations. One activating mutation, p.Pro252Arg in the FGFR1 gene, accounts for approximately 5 % of the diagnosed patients. Mutations in FGFR2 are found in the IgIIIa and IgIIIc regions as well as the tyrosine kinase domain, and tend to be phenotypically more severe than the FGFR1-derived phenotype.
Crouzon Syndrome With or Without Acanthosis Nigricans
Crouzon syndrome (OMIM#123500) patients have multiple suture fusions or coronal fusions causing brachycephaly, trigonocephaly, and rare reports of cloverleaf skull malformation also known as kleeblattschädel [53, 54]. Attributes typically include hypertelorism, a small midface, beaked nose and protrusion of the eyes. Hands and feet are generally normal. Heterozygous mutations in the FGFR2 gene show high penetrance and variability of the phenotype within families. Approximately half of the cases are inherited and the other half arise from de novo mutations. Increasing paternal age is a contributing factor for de novo mutations [35]. The prevalence is estimated as 1 in 65,000 live births [55].
Patients with Crouzon syndrome with acanthosis nigricans (OMIM#612247) are typically female, display hyperpigmentation of the skin, hyperkeratosis, and other skin findings. A specific FGFR3 heterozygous mutation, p.Ala391Glu, has been identified and is located in the transmembrane domain [56].
Muenke Syndrome
Muenke syndrome (OMIM#602849) displays incomplete penetrance and a variable phenotype even within families [57]. Prevalence in the population is estimated to be 1 in 30,000 live births. Sporadic and familial cases have been reported. Characteristics include bi- or unicoronal synostosis, midfacial hypoplasia, ptosis, and downslanting palpebral fissures. Some affected individuals have additional features that may include sensorineural hearing loss, developmental delay, brachydactyly, and coned epiphyses in the hands and feet. Muenke craniosynostosis is a result of a specific heterozygous mutation, p.Pro250Arg, found in the linker region between domains IgII and IgIII of FGFR3 [58–61]. Targeted testing may be performed by sequencing the seventh exon of FGFR3 or by RFLP analysis using the MspI endonuclease.
Beare-Stevenson Syndrome
Beare-Stevenson (OMIM#123790), also known as cutis gyrate syndrome of Beare and Stevenson, is a rare and severe disorder. Patients characteristically have body-wide skin furrows (cutis gyrate), acanthosis nigricans, skin tags, bifid scrotum, and anogenital anomalies. The craniosynostosis may be severe and present as a cloverleaf skull with hypertelorism, a broad nasal bridge, cleft palate, and hypodontia. Two heterozygous point mutations in the FGFR2 gene, p.Ser372Cys and p.Tyr375Cys, account for 50–60 % of cases, suggesting locus heterogeneity. The resulting cysteine residues are thought to increase ligand-independent dimerization. These de novo mutations are analogous to the mutations in FGFR3 causing thanatophoric dysplasia. An intragenic deletion, c.1506del63, has recently been described and is proposed to alter gene splicing in favor of the IgIIIb isoform of FGFR2. Loss of the 21 amino acids encoded by exons 8 and 9 is suggested to cause aberrant expression of FGFR2b [62].
Jackson-Weiss Syndrome
Jackson-Weiss Syndrome (OMIM#123150), inherited in an autosomal dominant manner, has been most prominently described in an extended Amish family with a p.Ala344Gly mutation in the IgIIIc domain of FGFR2 [63–66]. Fully penetrant with variable severity, the characteristics of the syndrome include craniosynostosis with facial anomalies, broad great toes, and webbing of the second and third toes [67]. A few reports suggest mutations in FGFR2 and FGFR1 exhibit phenotypic traits similar to Jackson-Weiss syndrome indicating that Crouzon, Jackson-Weiss, and Pfeiffer syndromes may represent a spectrum of craniosynostotic and digit malformations [68, 69].
Antley-Bixler Syndrome Type 2
Antley-Bixler syndrome (ABS; trapezoidocephaly-synostosis syndrome) is a rare and severe heterogeneous disorder with mutations found in both the FGFR2 gene (autosomal dominant; type 2; OMIM#207410) and the cytochrome P450 oxidoreductase (POR) gene (autosomal recessive; type 1; OMIM#201750). ABS type 2 is characterized by craniosynostosis of the coronal and lambdoid sutures, midfacial hypoplasia, radiohumeral and digit fusions, exophthalmos, and arachnodactyly [70, 71]. The mutations associated with ABS include p.Trp290Cys and p.Ser351Cys, both found in the IgIII domain of the FGFR2 gene [72]. Mutations at these positions also have been associated with the milder phenotype of Crouzon syndrome [73].
Osteoglophonic Dysplasia
Osteoglophonic dysplasia (Fairbank-Keats syndrome; OMIM#166250), a very rare disorder, is typified by variable craniosynostosis and rhizomelic dwarfism with a “hollowed-out” appearance of the tubular bones on radiographs, depression of the nasal bridge, unerupted teeth, frontal bossing, and prognathism similar to achondroplasia. Mutations in FGFR1 are found in the conserved amino acids clustered in the C-terminal region of the IgIII immunoglobulin domain, the linker region, and the transmembrane domain. The FGFR1 p.Tyr372Cys mutation is analogous to both the FGFR2 p.Tyr375Cys mutation that causes Beare-Stevenson syndrome and the p.Tyr373Cys FGFR3 mutation that causes thanatophoric dwarfism type 1, indicating the importance of that amino acid position in the functional role of the receptors [74].
Achondroplasia
Achondroplasia (OMIM#100800) arises from mutations in the FGFR3 gene that inhibit chondrocyte proliferation within the endochondral growth plate resulting in the shortening of long bones. Achondroplasia is the most common form of FGFR-related short-limbed dwarfism [75, 76], with an occurrence of 1 in 10,000 to 30,000 live births [4, 5]. Two common variants, c.1138G>A (~98 %) and c.1138G>C (1–2 %), result in a p.Gly380Arg mutation in the transmembrane domain of FGFR3 [77]. The achondroplasia mutation is the most common de novo disease-causing mutation known. There is a strong paternal origin for the mutation, mostly in fathers over the age of 35 years [34]. A second mutation, described in several published accounts, suggests that p.Gly375Cys also causes achondroplasia [78–80].
Testing for the c.1138G>A mutation may be performed by RFLP digestion of exon 10 with the SfcI restriction enzyme. It has been noted, however, that complete digestion is not consistently observed for the assay and other molecular methods may be required to differentiate between the G>A and the G>C mutations, the heterozygous form, and the lethal homozygous form [81–83].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


