Environmental toxicology is the study of deleterious effects of physical, chemical, or microbiological agents present in air, water, food, or other media. In this context, airborne exposures include those received by cigarette smokers and people working in various industries, as well as those received by all of us from pollutants. Many of the principles and mechanisms pertinent to drug toxicity, discussed in Chapter 6, Drug Toxicity, apply also to nondrug toxicants. In particular, the tenet of dose–response explains why low-level exposures to ubiquitous chemicals are typically harmless, while increasing levels of exposure confer increased risks of harm.
In the United States and elsewhere, actions by regulatory agencies such as the US Food and Drug Administration (FDA), the Occupational Safety and Health Administration, and the Environmental Protection Agency have resulted in foods, workplaces, and environments that are significantly safer than they were in the mid-twentieth century and earlier. Nonetheless, accidental poisonings, food poisoning, cigarette smoking, excessive consumption of alcoholic beverages, and the legacy of occupational overexposures to asbestos, silica, and other occupational carcinogens continue to be responsible for considerable burdens of disease. Similarly, although gross overexposures of children and others to the toxic metal lead are becoming less common in much of the world (due primarily to the removal of tetraethyl lead from gasoline and the reduced use of lead-based pigments), environmental sources of lead remain, and children deficient in iron and calcium in particular are at risk of lead-induced neurobehavioral disease. Worldwide, the prevalence of, and protections against, toxic agents vary widely, both across and within countries, such that the health of children, workers, and others may be considerably compromised. This is especially the case in groups suffering from malnutrition, chronic infections, and other insults that, per se and in concert with toxic exposures, harm health.
 The W family is running out of money. Mr. W has lost his job, and Ms. W’s hours have been cut back. After months of trying to make ends meet, Mr. W decides to stop paying the electricity bill. He borrows a propane generator from a friend and sets it up in the garage attached to the house. That evening, Mr. W, his wife, and their teenage son dine at home. Ms. W feels as if she is coming down with the flu, Mr. W has a headache, and the son feels irritable. All three retire early to bed.
The W family is running out of money. Mr. W has lost his job, and Ms. W’s hours have been cut back. After months of trying to make ends meet, Mr. W decides to stop paying the electricity bill. He borrows a propane generator from a friend and sets it up in the garage attached to the house. That evening, Mr. W, his wife, and their teenage son dine at home. Ms. W feels as if she is coming down with the flu, Mr. W has a headache, and the son feels irritable. All three retire early to bed.
The next morning, the son is absent from school, and Ms. W fails to arrive at work. Friends call, but the phone goes unanswered. The police are notified; they arrive, break in, and find all three dead in their beds.
Questions
1. What toxic agent(s) could have caused the family members’ deaths?
2. Which routine laboratory test(s) could confirm the likely cause of death?
 ACUTE AND SUBCHRONIC TOXICOLOGY
ACUTE AND SUBCHRONIC TOXICOLOGY
Numerous substances can cause serious acute illness and death. This section describes some common causes of acute and subchronic poisoning, the mechanisms by which they act, and, as appropriate, treatments.
The combustion of any organic material produces carbon monoxide (CO) gas and other products of incomplete combustion. Insufficiently vented home-heating furnaces, wood stoves, and other combustion sources can result in accumulation of CO in indoor air to toxic concentrations. The propane generator in the introductory case was not sufficiently ventilated, causing carbon monoxide to reach lethal concentrations. In the United States, some 15,000 emergency department visits and 500 deaths annually are caused by overexposures to CO. These deaths are exclusive of fire-related deaths, many of which are also caused in part by elevated concentrations of CO.
CO causes tissue hypoxia by binding more than 200-fold more tightly to the heme iron in hemoglobin than does O2, thereby reducing the transport of oxygen in the blood (Fig. 50-1). In addition, carboxyhemoglobin (COHb) shifts the dissociation curve for oxyhemoglobin (OHb) to the left, impeding the dissociation of O2. CO also binds to cytochromes and to myoglobin in heart and skeletal muscle; this bound CO can serve as an internal reservoir of CO as COHb concentrations in the blood decrease. Binding of CO to myoglobin in the myocardium interferes with oxidative phosphorylation, thus depriving heart muscle of energy. Patients with cardiac disorders are particularly susceptible to acute CO poisoning, and survivors of moderate to severe CO poisoning appear to be at increased risk of cardiac death years after the incident.
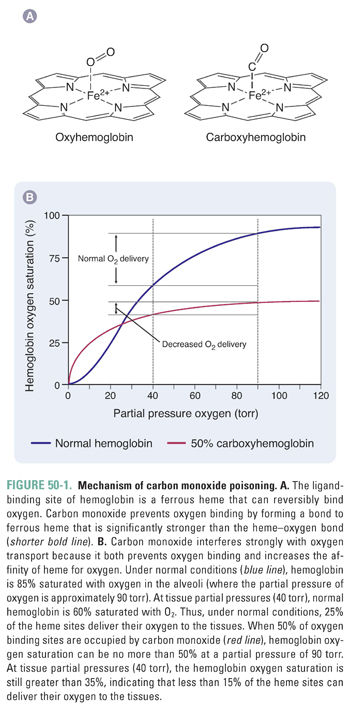
Because the initial symptoms of CO poisoning are nonspecific, including headache, dizziness, and irritability, an accurate diagnosis is sometimes elusive. The W family did not react to the presence of carbon monoxide because it is odorless and nonirritating and because the symptoms they experienced shortly before sleep would not have been a cause of alarm. Had their house been equipped with a carbon monoxide detector, their deaths would almost certainly have been prevented. Measurement of COHb is straightforward, and concentrations higher than about 3% in nonsmokers and 5–10% in smokers indicate an unusual exposure. (Note that pO2, the partial pressure of oxygen, is likely to be normal in a CO-poisoned patient.) Signs and symptoms of acute poisoning track approximately with COHb concentrations, with severe headache, vomiting, and visual disturbances at 30–40% COHb and collapse and convulsions at 50–60% COHb. Death is likely at 70% COHb and possible at lower concentrations.
Survivors of severe CO poisoning are at risk of brain damage; areas of the brain with high oxygen demand are most likely to be impaired, although the mechanisms and outcomes of CO poisoning differ from those of simple hypoxia. CO-induced neuropathy is due to marked cerebral vasodilation, mitochondrial dysfunction, cell death from apoptosis, and, upon reoxygenation, reperfusion injury.
The half-life of COHb is approximately 5 hours in room air and decreases to about 90 minutes in a 100% O2 environment at normal pressure. Hyperbaric oxygen therapy (3 atmospheres, 100% O2) can reduce the half-life to about 20 minutes and appears to protect against long-term brain damage by improving energy metabolism, minimizing lipid peroxidation, and decreasing neutrophil adherence. A rule of thumb is that victims with COHb concentrations above 25% (or above 15% in pregnant women) should receive hyperbaric oxygen therapy. However, COHb concentrations alone are only approximate indicators of risk, and use of hyperbaric oxygen therapy is preferred when available.
The cyanide ion (C≡N−) is a highly toxic and frequently lethal poison. It may be inhaled, ingested, or absorbed through the skin from sources as diverse as hydrogen cyanide gas, cyanide salts, apricot pits, peach pits, cherry pits, cassava, fire smoke, and vapors from industrial metal plating operations. Cyanide is also a metabolite of nitriles and nitroprusside. Cyanide binds to the ferric iron in the heme a3/CuB center of cytochrome c oxidase, thereby blocking aerobic respiration and preventing cellular use of oxygen. This causes a shift to anaerobic metabolism and a resulting metabolic acidosis. As with CO poisoning, cyanide poisoning damages tissues with high oxygen demand, such as the brain and heart.
Signs and symptoms of cyanide poisoning depend on dose and route of exposure and are somewhat nonspecific: headache, confusion, altered mental status, hypertension (early) or hypotension (late), nausea, and other symptoms are all possible. Pallor or cyanosis is not present (assuming that there is no co-exposure to carbon monoxide). Unless cyanide exposure is reported or witnessed, or is likely to have occurred given the patient’s occupation or recent activities, diagnosis may be difficult. Sometimes, an odor of bitter almonds may be noted. Because cyanide is cleared rapidly from the blood, and because of technical challenges, measurements of cyanide in the blood may be both time-consuming and misleading. Moreover, some endogenous production of cyanide occurs in healthy individuals, and smokers’ blood contains elevated cyanide concentrations. There is some debate about the blood concentrations of cyanide deemed toxic or potentially lethal, but 1 mg/L (39 μmol/L) is typically regarded as a potentially toxic level.
Treatment for acute cyanide poisoning may include decontamination, supportive therapy, and administration of an antidote. Decontamination may entail simply the removal of contaminated clothes, and care should be taken to avoid inadvertent exposure of responders to the cyanide-containing material. Supportive therapy, including supplemental oxygen, should aim to avoid organ failure and may be needed to address toxicities such as coma, lactic acidosis, hypotension, and respiratory failure.
The traditional antidotal treatment for acute cyanide poisoning in the United States is a cyanide antidote “kit” (CAK) that contains amyl nitrite, sodium nitrite, and sodium thiosulfate. The nitrites act by oxidizing hemoglobin to methemoglobin to provide a substrate that can compete with heme a3 in cytochrome c oxidase for cyanide molecules. Amyl nitrite is usually given by inhalation and acts (and is cleared from the bloodstream) rapidly, while sodium nitrite is administered intravenously and has a longer duration of action. The methemoglobin-bound cyanide is oxidized to the relatively nontoxic thiocyanate by the enzyme rhodanese (also known as transsulfurase) and excreted in urine. Sodium thiosulfate provides a ready source of sulfur for the detoxication reaction and enhances cyanide metabolism.
Importantly, use of the CAK may pose significant risk to the patient, since a substantial fraction of hemoglobin must be converted to methemoglobin to compete effectively for the cyanide ion. Smoke inhalation victims (who have high levels of exposure to carbon monoxide) are sometimes presumed to have been poisoned by cyanide gas. Such patients may already be suffering from hypoxia before CAK treatment is begun. Exacerbation of hypoxia by forcing conversion of hemoglobin to methemoglobin may be detrimental to such patients. CAK should also be avoided in pregnant women and infants, who may carry fetal hemoglobin and have immature methemoglobin reductase activity. Furthermore, the CAK may cause severe hypotension and lead to cardiovascular collapse.
Concerns regarding possible terrorist use of cyanide led to the approval in 2006 by the FDA of an alternative antidote, hydroxocobalamin (Cyanokit®). This member of the vitamin B12 family is an endogenous compound that was already in use at lower doses for treatment of vitamin B12 deficiency. The mechanism of action of this drug differs from that of the compounds in the CAK: the cobalt moiety in hydroxocobalamin has high affinity for cyanide and competes directly with the ferric iron in cytochrome c oxidase for cyanide, forming nontoxic cyanocobalamin that is excreted in the urine. Hydroxocobalamin is generally well tolerated, but anaphylactic reactions are possible. The compound also causes urine to have a bright red color for about a week and may discolor the skin at the site of injection. Interference with spectrophotometric tests and assays for oxyhemoglobin, carboxyhemoglobin, and methemoglobin may also occur.
Lead is ubiquitous in the environment because of its persistence, its formerly widespread and unnecessary use as a gasoline additive, and its use in pigments, paints, plumbing, solder, and other products. Lead is toxic to the central nervous system, making exposure a particular concern for fetuses and children up to the age of about 7 years. Young children are also at risk because they are more likely than adults to ingest lead-contaminated paint dust and other nonfood materials. Despite a fivefold decrease in the exposure to lead in the United States and elsewhere since the mid-twentieth century, children today may still be at risk of developing lead-induced neurocognitive deficits. This is especially true for children who live near active, poorly controlled lead mines or smelters or in countries where leaded fuels are, or recently were, used. (Leaded gasoline was not banned in China until 2000, for example.) Lead-glazed clay cookware and solder remain common in some areas, and some of this lead contaminates food and water. Exposures to lead that present no overt symptoms may nonetheless be toxic, and testing young children’s blood lead levels is essential. Although the half-life of lead in soft tissues is relatively short, its half-life in bone is more than 20 years; a substantial exposure in early childhood can result in elevated bone lead levels for decades.
Lead disrupts the blood–brain barrier, allowing both lead and other potential neurotoxins to reach the CNS. There, lead can block voltage-dependent calcium channels, interfere with neurotransmitter function, and, most importantly, interfere with cell–cell interactions in the brain; the latter effect causes permanent changes in neuronal circuitry. Overt lead encephalopathy, which is rare in the United States today, results in lethargy, vomiting, irritability, and dizziness and can progress to altered mental status, coma, and death. Low- and moderate-level exposures to young children are believed to result in IQ deficits of two to four points for every 10 μg/dL increase in blood lead concentration. Whether some blood lead levels are so low as to present essentially no risk of neurobehavioral deficit is a matter of debate and ongoing research.
Lead interferes with the synthesis of hemoglobin at multiple steps and thereby causes microcytic, hypochromic anemia. Specifically, lead inhibits delta-aminolevulinic acid dehydratase (ALA-D), which catalyzes the synthesis of porphobilinogen, a heme precursor. Lead also inhibits the incorporation of iron into the porphyrin ring.
In the kidney, lead causes both reversible and irreversible toxicity. Lead can interfere reversibly with energy production in proximal tubular cells by interfering with mitochondrial function, resulting in decreased energy-dependent reabsorption of ions, glucose, and amino acids. Chronic exposure to lead results in interstitial nephritis, with the eventual development of fibrosis and chronic kidney disease.
When indicated clinically, body burdens of metals such as lead, mercury, or cadmium can be reduced using electron donors such as an amine, hydroxide, carboxylate, or mercaptan to form metal–ligand complexes. A chelator, which in Greek means “claw,” is a multidentate structure with multiple binding sites (Fig. 50-2). Binding of the metal at multiple sites shifts the equilibrium constant in favor of metal ligation. High-affinity metal–ligand binding is critical because the chelator must compete with tissue macromolecules for binding. In addition, the chelator should be nontoxic and water-soluble, and the complex should be readily cleared. Finally, an ideal chelator should have a low binding affinity for endogenous ions such as calcium. To prevent the depletion of tissue calcium, many chelators are administered as calcium complexes. The target metal is then exchanged for calcium, and the body’s calcium stores are not depleted.
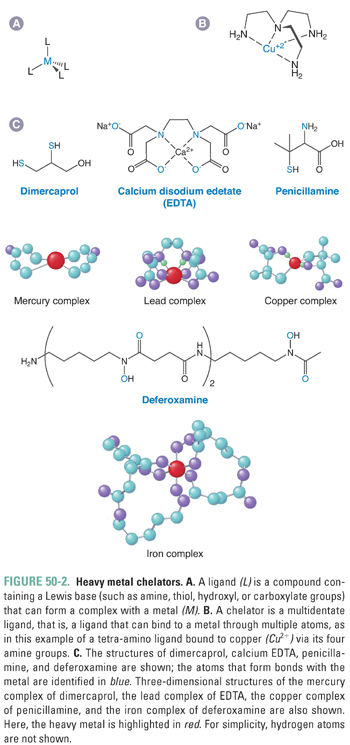
The most important heavy metal chelators are edetate disodium (the calcium, disodium complex of EDTA), which can be used to bind lead; dimercaprol (also known as British anti-Lewisite or BAL), which binds gold, arsenic, lead, and mercury to its two thiol groups; and succimer (2,3-dimercaptosuccinic acid), which has supplanted dimercaprol for the removal of lead, cadmium, mercury, and arsenic. Deferoxamine is used for the removal of toxic levels of iron, such as would occur in accidental overdoses of iron-containing supplements or in patients with transfusion-dependent anemias. Deferasirox is an orally bioavailable iron chelator that may supplant deferoxamine for many conditions associated with chronic iron overload. Removal of copper, typically in patients with Wilson’s disease, is accomplished with penicillamine or, for patients who do not tolerate penicillamine, trientine.
An estimated one in four Americans experience significant food-borne illnesses each year. The mechanisms of food poisoning involve either infection, which typically manifests one to several days after exposure, or intoxication from a preformed microbial or algal toxin, with symptoms occurring within a few hours of exposure. Infectious food poisoning is typically caused by species of Salmonella, Listeria, Cryptosporidium, or Campylobacter. Less common but quite virulent are poisonings by enteropathogenic Escherichia coli, which can cause sometimes-fatal hemorrhagic colitis and hemolytic uremic syndrome (HUS), likely through the uptake of pathologic bacterial proteins by host cells.
Food intoxication is often caused by toxins elaborated by Staphylococcus aureus or Bacillus cereus or by marine algal toxins ingested via seafood. S. aureus produces a variety of toxins; the staphylococcal enterotoxins (SE) induce emesis by stimulating receptors in the abdominal viscera. High-protein foods, such as meats, cold cuts, and egg and dairy products, are contaminated with S. aureus upon improper food handling after cooking, followed by poor refrigeration.
B. cereus, a common contaminant of cooked rice, produces several toxins that cause vomiting and diarrhea. Of particular concern is the production of cereulide, a small, cyclic peptide that stimulates intestinal 5-HT3 receptors, resulting in emesis. The peptide is heat-stable to 259°F for up to 90 minutes, so reheating of contaminated cooked rice will typically not prevent intoxication.
Most algal toxins are neurotoxic and heat-stable, so, again, cooking leaves the toxins intact. Algal saxitoxins are a group of approximately 20 heterocyclic guanidine derivatives that bind with high affinity to the voltage-gated sodium channel, thus inhibiting neuronal activity and causing tingling and numbness, loss of motor control, drowsiness, incoherence, and, with sufficient doses (greater than about 1 mg), respiratory paralysis.
Many food-borne illnesses appear to be caused by pathogens that are not yet characterized. Moreover, novel pathogens can emerge because of changing ecologies or technologies or can arise via transfer of mobile virulence factors such as bacteriophages.
Acute illness can also be caused by mistaken ingestion of nonfood items, such as poisonous mushrooms collected by amateur mycologists or any number of poisonous plants. The highly toxic “death cap” mushroom, for instance, Amanita phalloides, produces numerous cyclopeptide toxins that are not destroyed by cooking or drying, have no distinctive taste, and are taken up by hepatocytes. The amatoxins bind tightly to RNA polymerase II, substantially slowing RNA and protein synthesis and leading to hepatocyte necrosis. The somewhat less toxic phallotoxins and virotoxins interfere with F- and G-actins in the cytoskeleton. Consumption of Amanita species or their relatives can thus cause severe liver dysfunction, hepatic and renal failure, and death. Initial symptoms of poisoning, such as abdominal pain, nausea, severe vomiting and diarrhea, fever, and tachycardia, may occur 6–24 hours after consumption of the mushrooms. Hepatic and renal function may deteriorate even while the initial symptoms abate, leading to jaundice, hepatic encephalopathy, and fulminant liver failure; death may occur 4–9 days after consumption. There is no specific antidote.
An anticholinergic syndrome may be caused by deliberate or accidental ingestion of jimson weed, a plant belonging to the Datura family. All parts of the plant are toxic, but the seeds and leaves, in particular, contain atropine, scopolamine, and hyoscyamine. These compounds are rapidly absorbed and produce anticholinergic symptoms such as mydriasis, dry, flushed skin, agitation, tachycardia, hyperthermia, and hallucinations. The mnemonic for anticholinergic effects, “blind as a bat, dry as a bone, red as a beet, mad as a hatter, and hot as a hare,” is applicable to jimson weed poisoning.
Some plants in the families Umbelliferae (such as parsley, parsnip, dill, celery, and giant hogweed), Rutaceae (such as limes and lemons), and Moraceae (such as figs) contain psoralen isomers (furocoumarins) in leaves, stems, or sap that can be absorbed into the skin after contact. Subsequent exposure to ultraviolet (UV) A radiation of wavelength >320 nm (generally via sunlight) can excite the furocoumarins, resulting in epidermal tissue damage. Within 2 days, burning, redness, and blistering are observed in areas of contact with the plant and light; after healing, hyperpigmentation may persist for months. The response is greater with increasing plant contact, humidity, and duration and intensity of radiation exposure. This nonallergic phytophototoxic mechanism is the basis of psoralen + UV-A (PUVA) therapy for eczema and other dermatologic disorders.
Strong acids, alkalis (caustic agents), oxidants, and reducing agents damage tissue by altering the structure of proteins, lipids, carbohydrates, and nucleic acids so severely that cellular integrity is lost. These substances, such as potassium hydroxide in drain cleaners and sulfuric acid in car batteries, produce chemical burns by hydrolyzing, oxidizing, or reducing biological macromolecules or by denaturing proteins. High concentrations of detergents can also cause nonspecific tissue damage by disrupting and dissolving the plasma membrane of cells.
Although some of these agents may target particular macromolecules, direct tissue-damaging agents tend to be relatively nonspecific. Thus, the systems most commonly affected are those most exposed to the environment. Skin and eyes are frequently affected by splashes or spills. The respiratory system is affected when toxic gases or vapors are inhaled, and the digestive system is affected by accidental or deliberate ingestion of toxic substances.
Many agents can cause damage to deep tissues after breaking through the barrier formed by the skin. Other agents are able to pass through the skin while causing relatively little local damage but destroy deeper tissues such as muscle or bone. For example, hydrofluoric acid (HF; found in, among other products, grout cleaner) causes milder skin burns than an equivalent amount of hydrochloric acid (HCl). However, once HF reaches deeper tissue, it destroys the calcified matrix of bone. In addition to the direct effects of the acid, the release of calcium stored in bone can cause life-threatening cardiac arrhythmias. For this reason, HF can be more dangerous than an equivalent amount of HCl.
Three characteristics determine the extent of tissue damage: the compound’s identity, its concentration/strength, and its buffering capacity, or its ability to resist change in pH or redox potential. As mentioned above, HF is more injurious than an equivalent amount of HCl. In general, a stronger acid or base (measured by pH) or oxidant or reductant (measured by redox potential) will cause more damage than an equivalent compound at a more physiologic pH or redox potential. A solution of 10−2 M sodium hydroxide in water has a pH of 12 but has low capacity to cause tissue damage because it has a small buffering capacity and is rapidly neutralized by body tissue. In contrast, a buffered solution of pH 12, such as that found in wet ready-set concrete [made with buffered Ca(OH)2], can cause more serious alkali burns because tissues cannot readily neutralize the material’s extreme pH.
Pesticides include insecticides, herbicides, rodenticides, and other compounds designed to kill unwanted organisms in the environment. By their nature, pesticides—of which there are hundreds (vastly more natural than synthetic)—are biologically active; however, the degree of their specificity toward target organisms varies, and many of these compounds cause toxicity in humans and other nontarget organisms. Some of the more common acute poisonings involve organophosphate and pyrethroid insecticides and rodenticides.
Organophosphate insecticides, derived from phosphoric or thiophosphoric acid, include parathion, malathion, diazinon, fenthion, chlorpyrifos, and many other chemicals. These widely used compounds are acetylcholinesterase (AChE) inhibitors due to their ability to phosphorylate AChE at its esteratic active site (Fig. 50-3). Inhibition of AChE, and consequent accumulation of acetylcholine at cholinergic junctions in nerve tissue and effector organs, produces acute muscarinic, nicotinic, and central nervous system (CNS) effects such as bronchoconstriction, increased bronchial secretions, salivation, lacrimation, sweating, nausea, vomiting, diarrhea, and miosis (muscarinic signs), as well as twitching, fasciculations, muscle weakness, cyanosis, and elevated blood pressure (nicotinic signs). CNS effects can include anxiety, restlessness, confusion, and headache. Symptoms usually occur within minutes or hours of exposure and resolve within a few days in nonlethal poisonings.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


