62
Drugs Used in the Treatment of Gastrointestinal Diseases
CASE STUDY
A 21-year-old woman comes with her parents to discuss therapeutic options for her Crohn’s disease. She was diagnosed with Crohn’s disease 2 years ago, and it involves her terminal ileum and proximal colon, as confirmed by colonoscopy and small bowel radiography. She was initially treated with mesalamine and budesonide with good response, but over the last 2 months, she has had a relapse of her symptoms. She is experiencing fatigue, cramping, abdominal pains, and nonbloody diarrhea up to 10 times daily, and she has had a 15-lb weight loss.
She has no other significant medical or surgical history. Her current medications are mesalamine 2.4 g/d and budesonide 9 mg/d. She appears thin and tired. Abdominal examination reveals tenderness without guarding in the right lower quadrant; no masses are palpable. On perianal examination, there is no tenderness, fissure, or fistula. Her laboratory data are notable for anemia and elevated C-reactive protein. What are the options for immediate control of her symptoms and disease? What are the long-term management options?
INTRODUCTION
Many of the drug groups discussed elsewhere in this book have important applications in the treatment of diseases of the gastrointestinal tract and other organs. Other groups are used almost exclusively for their effects on the gut; these are discussed in the following text according to their therapeutic uses.
 DRUGS USED IN ACID-PEPTIC DISEASES
DRUGS USED IN ACID-PEPTIC DISEASES
Acid-peptic diseases include gastroesophageal reflux, peptic ulcer (gastric and duodenal), and stress-related mucosal injury. In all these conditions, mucosal erosions or ulceration arise when the caustic effects of aggressive factors (acid, pepsin, bile) overwhelm the defensive factors of the gastrointestinal mucosa (mucus and bicarbonate secretion, prostaglandins, blood flow, and the processes of restitution and regeneration after cellular injury). Over 90% of peptic ulcers are caused by infection with the bacterium Helicobacter pylori or by use of nonsteroidal anti-inflammatory drugs (NSAIDs). Drugs used in the treatment of acid-peptic disorders may be divided into two classes: agents that reduce intragastric acidity and agents that promote mucosal defense.
AGENTS THAT REDUCE INTRAGASTRIC ACIDITY
PHYSIOLOGY OF ACID SECRETION
The parietal cell contains receptors for gastrin (CCK-B), histamine (H2), and acetylcholine (muscarinic, M3) (Figure 62–1). When acetylcholine (from vagal postganglionic nerves) or gastrin (released from antral G cells into the blood) bind to the parietal cell receptors, they cause an increase in cytosolic calcium, which in turn stimulates protein kinases that stimulate acid secretion from a H+/K+-ATPase (the proton pump) on the canalicular surface.
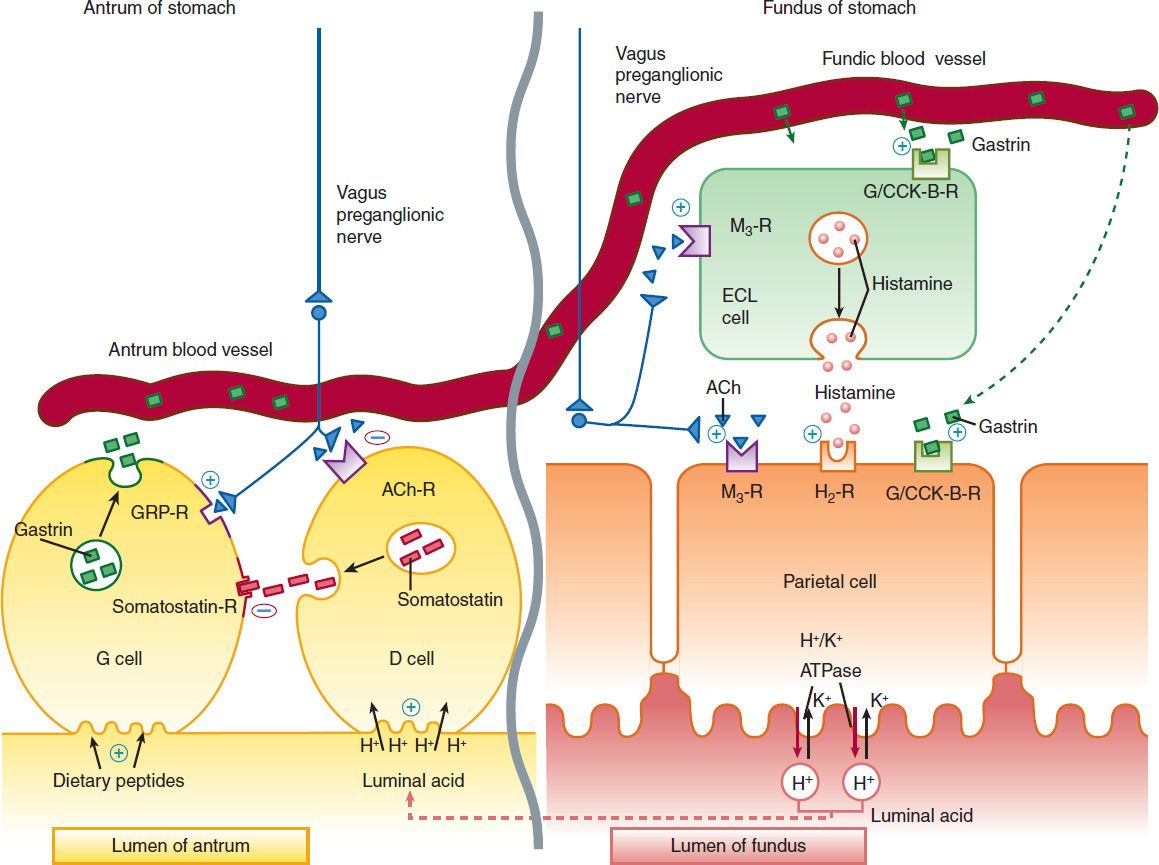
FIGURE 62–1 Schematic model for physiologic control of hydrogen ion (acid) secretion by the parietal cells of the gastric fundic glands. Parietal cells are stimulated to secrete acid (H+) by gastrin (acting on gastrin/CCK-B receptor), acetylcholine (M3 receptor), and histamine (H2 receptor). Acid is secreted across the parietal cell canalicular membrane by the H+/K+-ATPase proton pump into the gastric lumen. Gastrin is secreted by antral G cells into blood vessels in response to intraluminal dietary peptides. Within the gastric body, gastrin passes from the blood vessels into the submucosal tissue of the fundic glands, where it binds to gastrin-CCK-B receptors on parietal cells and enterochromaffin-like (ECL) cells. The vagus nerve stimulates postganglionic neurons of the enteric nervous system to release acetylcholine (ACh), which binds to M3 receptors on parietal cells and ECL cells. Stimulation of ECL cells by gastrin (CCK-B receptor) or acetylcholine (M3 receptor) stimulates release of histamine. Within the gastric antrum, vagal stimulation of postganglionic enteric neurons enhances gastrin release directly by stimulation of antral G cells (through gastrin-releasing peptide, GRP) and indirectly by inhibition of somatostatin secretion from antral D cells. Acid secretion must eventually be turned off. Antral D cells are stimulated to release somatostatin by the rise in intraluminal H+ concentration and by CCK that is released into the bloodstream by duodenal I cells in response to proteins and fats (not shown). Binding of somatostatin to receptors on adjacent antral G cells inhibits further gastrin release. ATPase, H+/K+-ATPase proton pump; CCK, cholecystokinin; M3-R, muscarinic receptors.
In close proximity to the parietal cells are gut endocrine cells called enterochromaffin-like (ECL) cells. ECL cells also have receptors for gastrin and acetylcholine, which stimulate histamine release. Histamine binds to the H2 receptor on the parietal cell, resulting in activation of adenylyl cyclase, which increases intracellular cyclic adenosine monophosphate (cAMP) and activates protein kinases that stimulate acid secretion by the H+/K+-ATPase. In humans, it is believed that the major effect of gastrin upon acid secretion is mediated indirectly through the release of histamine from ECL cells rather than through direct parietal cell stimulation. In contrast, acetylcholine provides potent direct parietal cell stimulation.
ANTACIDS
Antacids have been used for centuries in the treatment of patients with dyspepsia and acid-peptic disorders. They were the mainstay of treatment for acid-peptic disorders until the advent of H2−receptor antagonists and proton-pump inhibitors (PPIs). They continue to be used commonly by patients as nonprescription remedies for the treatment of intermittent heartburn and dyspepsia.
Antacids are weak bases that react with gastric hydrochloric acid to form a salt and water. Their principal mechanism of action is reduction of intragastric acidity. After a meal, approximately 45 mEq/h of hydrochloric acid is secreted. A single dose of 156 mEq of antacid given 1 hour after a meal effectively neutralizes gastric acid for up to 2 hours. However, the acid-neutralization capacity among different proprietary formulations of antacids is highly variable, depending on their rate of dissolution (tablet versus liquid), water solubility, rate of reaction with acid, and rate of gastric emptying.
Sodium bicarbonate (eg, baking soda, Alka Seltzer) reacts rapidly with hydrochloric acid (HCl) to produce carbon dioxide and sodium chloride. Formation of carbon dioxide results in gastric distention and belching. Unreacted alkali is readily absorbed, potentially causing metabolic alkalosis when given in high doses or to patients with renal insufficiency. Sodium chloride absorption may exacerbate fluid retention in patients with heart failure, hypertension, and renal insufficiency. Calcium carbonate (eg, Tums, Os-Cal) is less soluble and reacts more slowly than sodium bicarbonate with HCl to form carbon dioxide and calcium chloride (CaCl2). Like sodium bicarbonate, calcium carbonate may cause belching or metabolic alkalosis. Calcium carbonate is used for a number of other indications apart from its antacid properties (see Chapter 42). Excessive doses of either sodium bicarbonate or calcium carbonate with calcium-containing dairy products can lead to hypercalcemia, renal insufficiency, and metabolic alkalosis (milk-alkali syndrome).
Formulations containing magnesium hydroxide or aluminum hydroxide react slowly with HCl to form magnesium chloride or aluminum chloride and water. Because no gas is generated, belching does not occur. Metabolic alkalosis is also uncommon because of the efficiency of the neutralization reaction. Because unabsorbed magnesium salts may cause an osmotic diarrhea and aluminum salts may cause constipation, these agents are commonly administered together in proprietary formulations (eg, Gelusil, Maalox, Mylanta) to minimize the impact on bowel function. Both magnesium and aluminum are absorbed and excreted by the kidneys. Hence, patients with renal insufficiency should not take these agents long-term.
All antacids may affect the absorption of other medications by binding the drug (reducing its absorption) or by increasing intragastric pH so that the drug’s dissolution or solubility (especially weakly basic or acidic drugs) is altered. Therefore, antacids should not be given within 2 hours of doses of tetracyclines, fluoroquinolones, itraconazole, and iron.
H2-RECEPTOR ANTAGONISTS
From their introduction in the 1970s until the early 1990s, H2-receptor antagonists (commonly referred to as H2 blockers) were the most commonly prescribed drugs in the world (see Clinical Uses). With the recognition of the role of H pylori in ulcer disease (which may be treated with appropriate antibacterial therapy) and the advent of PPIs, the use of prescription H2 blockers has declined markedly.
Chemistry & Pharmacokinetics
Four H2 antagonists are in clinical use: cimetidine, ranitidine, famotidine, and nizatidine. All four agents are rapidly absorbed from the intestine. Cimetidine, ranitidine, and famotidine undergo first-pass hepatic metabolism resulting in a bioavailability of approximately 50%. Nizatidine has little first-pass metabolism. The serum half-lives of the four agents range from 1.1 to 4 hours; however, duration of action depends on the dose given (Table 62–1). H2 antagonists are cleared by a combination of hepatic metabolism, glomerular filtration, and renal tubular secretion. Dose reduction is required in patients with moderate to severe renal (and possibly severe hepatic) insufficiency. In the elderly, there is a decline of up to 50% in drug clearance as well as a significant reduction in volume of distribution.
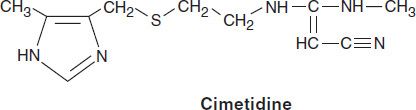
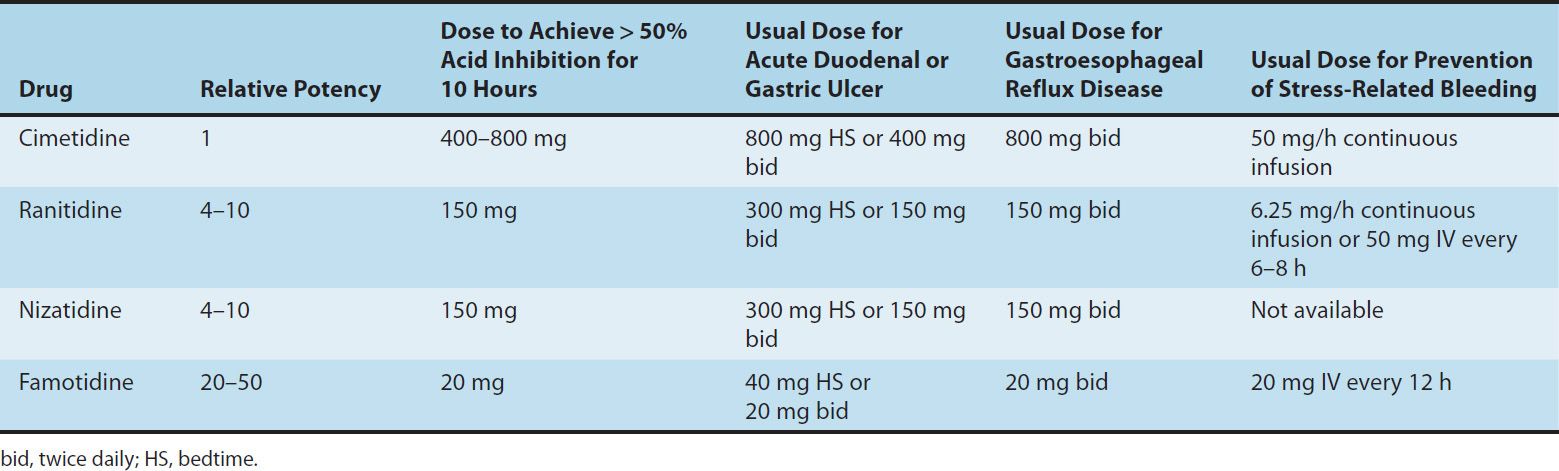
TABLE 62–1 Clinical comparisons of H2-receptor blockers.
Pharmacodynamics
The H2 antagonists exhibit competitive inhibition at the parietal cell H2 receptor and suppress basal and meal-stimulated acid secretion (Figure 62–2) in a linear, dose-dependent manner. They are highly selective and do not affect H1 or H3 receptors (see Chapter 16). The volume of gastric secretion and the concentration of pepsin are also reduced.
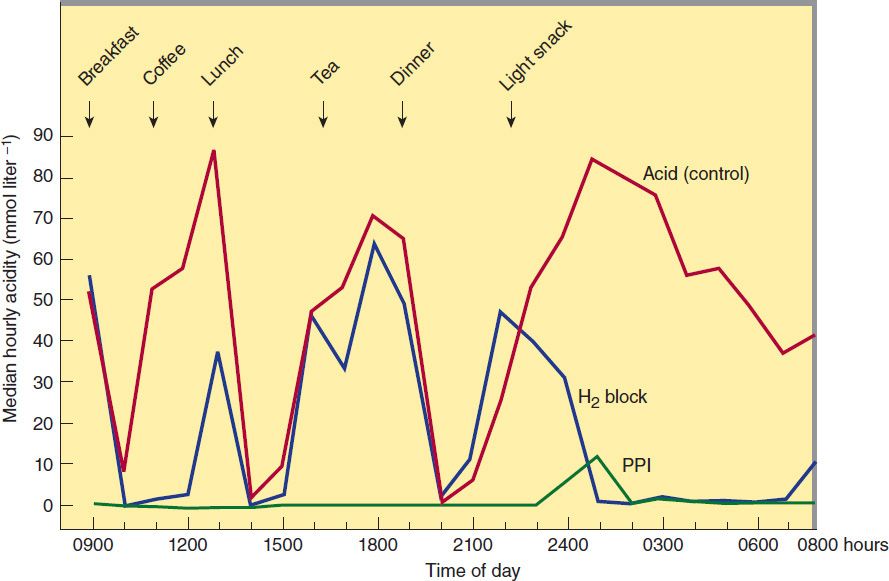
FIGURE 62–2 Twenty-four-hour median intragastric acidity pretreatment (red) and after 1 month of treatment with either ranitidine, 150 mg twice daily (blue, H2 block), or omeprazole, 20 mg once daily (green, PPI). Note that H2-receptor antagonists have a marked effect on nocturnal acid secretion but only a modest effect on meal-stimulated secretion. Proton pump inhibitors (PPIs) markedly suppress meal-stimulated and nocturnal acid secretion. (Data from Lanzon-Miller S et al: Twenty-four-hour intragastric acidity and plasma gastrin concentration before and during treatment with either ranitidine or omeprazole. Aliment Pharmacol Ther 1987;1:239.)
H2 antagonists reduce acid secretion stimulated by histamine as well as by gastrin and cholinomimetic agents through two mechanisms. First, histamine released from ECL cells by gastrin or vagal stimulation is blocked from binding to the parietal cell H2 receptor. Second, direct stimulation of the parietal cell by gastrin or acetylcholine has a diminished effect on acid secretion in the presence of H2-receptor blockade.
The potencies of the four H2-receptor antagonists vary over a 50-fold range (Table 62–1). When given in usual prescription doses however, all inhibit 60–70% of total 24-hour acid secretion. H2 antagonists are especially effective at inhibiting nocturnal acid secretion (which depends largely on histamine), but they have a modest impact on meal-stimulated acid secretion (which is stimulated by gastrin and acetylcholine as well as histamine). Therefore, nocturnal and fasting intragastric pH is raised to 4–5 but the impact on the daytime, meal-stimulated pH profile is less. Recommended prescription doses maintain greater than 50% acid inhibition for 10 hours; hence, these drugs are commonly given twice daily. At doses available in over-the-counter formulations, the duration of acid inhibition is less than 6 hours.
Clinical Uses
H2-receptor antagonists continue to be prescribed but PPIs (see below) are steadily replacing H2 antagonists for most clinical indications. However, the over-the-counter preparations of the H2 antagonists are heavily used by the public.
1. Gastroesophageal reflux disease (GERD)—Patients with infrequent heartburn or dyspepsia (fewer than 3 times per week) may take either antacids or intermittent H2 antagonists. Because antacids provide rapid acid neutralization, they afford faster symptom relief than H2 antagonists. However, the effect of antacids is short-lived (1–2 hours) compared with H2 antagonists (6–10 hours). H2 antagonists may be taken prophylactically before meals in an effort to reduce the likelihood of heartburn. Frequent heartburn is better treated with twice-daily H2 antagonists (Table 62–1) or PPIs. In patients with erosive esophagitis (approximately 50% of patients with GERD), H2 antagonists afford healing in less than 50% of patients; hence PPIs are preferred because of their superior acid inhibition.
2. Peptic ulcer disease—PPIs have largely replaced H2 antagonists in the treatment of acute peptic ulcer disease. Nevertheless, H2 antagonists are still sometimes used. Nocturnal acid suppression by H2 antagonists affords effective ulcer healing in most patients with uncomplicated gastric and duodenal ulcers. Hence, all the agents may be administered once daily at bedtime, resulting in ulcer healing rates of more than 80–90% after 6–8 weeks of therapy. For patients with ulcers caused by aspirin or other NSAIDs, the NSAID should be discontinued. If the NSAID must be continued for clinical reasons despite active ulceration, a PPI should be given instead of an H2 antagonist to more reliably promote ulcer healing. For patients with acute peptic ulcers caused by H pylori, H2 antagonists no longer play a significant therapeutic role. H pylori should be treated with a 10- to 14-day course of therapy including a PPI and two antibiotics (see below).
3. Nonulcer dyspepsia—H2 antagonists are commonly used as over-the-counter agents and prescription agents for treatment of intermittent dyspepsia not caused by peptic ulcer. However, benefit compared with placebo has never been convincingly demonstrated.
4. Prevention of bleeding from stress-related gastritis—Clinically important bleeding from upper gastrointestinal erosions or ulcers occurs in 1–5% of critically ill patients as a result of impaired mucosal defense mechanisms caused by poor perfusion. Although most critically ill patients have normal or decreased acid secretion, numerous studies have shown that agents that increase intragastric pH (H2 antagonists or PPIs) reduce the incidence of clinically significant bleeding. However, the optimal agent is uncertain at this time. For patients without a nasoenteric tube or with significant ileus, intravenous H2 antagonists are preferable over intravenous PPIs because of their proven efficacy and lower cost. Continuous infusions of H2 antagonists are generally preferred to bolus infusions because they achieve more consistent, sustained elevation of intragastric pH.
Adverse Effects
H2 antagonists are extremely safe drugs. Adverse effects occur in less than 3% of patients and include diarrhea, headache, fatigue, myalgias, and constipation. Some studies suggest that intravenous H2 antagonists (or PPIs) may increase the risk of nosocomial pneumonia in critically ill patients.
Mental status changes (confusion, hallucinations, agitation) may occur with administration of intravenous H2 antagonists, especially in patients in the intensive care unit who are elderly or who have renal or hepatic dysfunction. These events may be more common with cimetidine. Mental status changes rarely occur in ambulatory patients.
Cimetidine inhibits binding of dihydrotestosterone to androgen receptors, inhibits metabolism of estradiol, and increases serum prolactin levels. When used long-term or in high doses, it may cause gynecomastia or impotence in men and galactorrhea in women. These effects are specific to cimetidine and do not occur with the other H2 antagonists.
Although there are no known harmful effects on the fetus, H2 antagonists cross the placenta. Therefore, they should not be administered to pregnant women unless absolutely necessary. The H2 antagonists are secreted into breast milk and may therefore affect nursing infants.
H2 antagonists may rarely cause blood dyscrasias. Blockade of cardiac H2 receptors may cause bradycardia, but this is rarely of clinical significance. Rapid intravenous infusion may cause bradycardia and hypotension through blockade of cardiac H2 receptors; therefore, intravenous injections should be given over 30 minutes. H2 antagonists rarely cause reversible abnormalities in liver chemistry.
Drug Interactions
Cimetidine interferes with several important hepatic cytochrome P450 drug metabolism pathways, including those catalyzed by CYP1A2, CYP2C9, CYP2D6, and CYP3A4 (see Chapter 4). Hence, the half-lives of drugs metabolized by these pathways may be prolonged. Ranitidine binds 4–10 times less avidly than cimetidine to cytochrome P450. Negligible interaction occurs with nizatidine and famotidine.
H2 antagonists compete with creatinine and certain drugs (eg, procainamide) for renal tubular secretion. All of these agents except famotidine inhibit gastric first-pass metabolism of ethanol, especially in women. Although the importance of this is debated, increased bioavailability of ethanol could lead to increased blood ethanol levels.
PROTON-PUMP INHIBITORS (PPIS)
Since their introduction in the late 1980s, these efficacious acid inhibitory agents have assumed the major role for the treatment of acid-peptic disorders. PPIs are now among the most widely prescribed drugs worldwide due to their outstanding efficacy and safety.
Chemistry & Pharmacokinetics
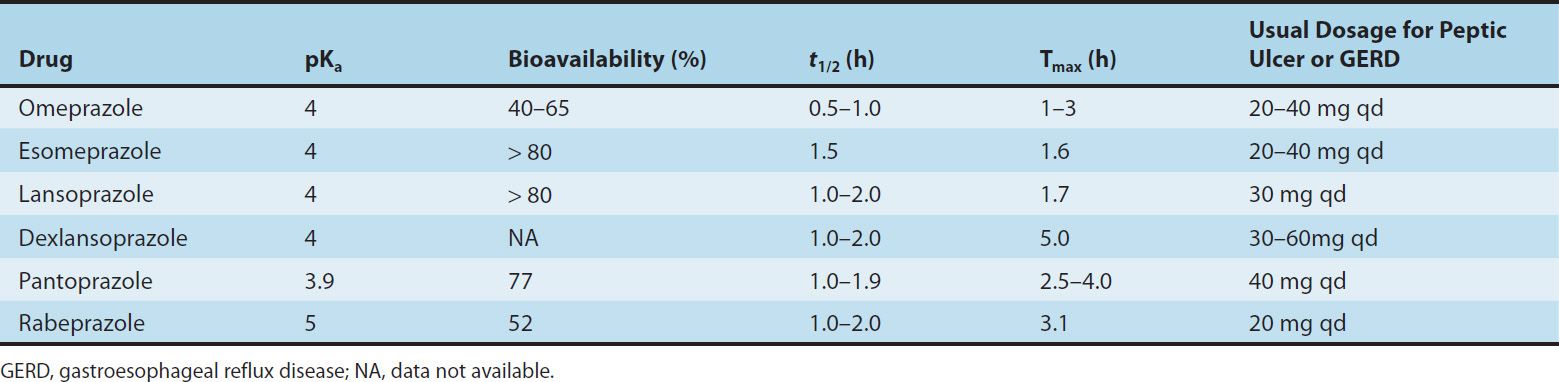
Six PPIs are available for clinical use: omeprazole, esomeprazole, lansoprazole, dexlansoprazole, rabeprazole, and pantoprazole. All are substituted benzimidazoles that resemble H2 antagonists in structure (Figure 62–3) but have a completely different mechanism of action. Omeprazole and lansoprazole are racemic mixtures of R– and S-isomers. Esomeprazole is the S-isomer of omeprazole and dexlansoprazole the R-isomer of lansoprazole. All are available in oral formulations. Esomeprazole and pantoprazole are also available in intravenous formulations (Table 62–2).
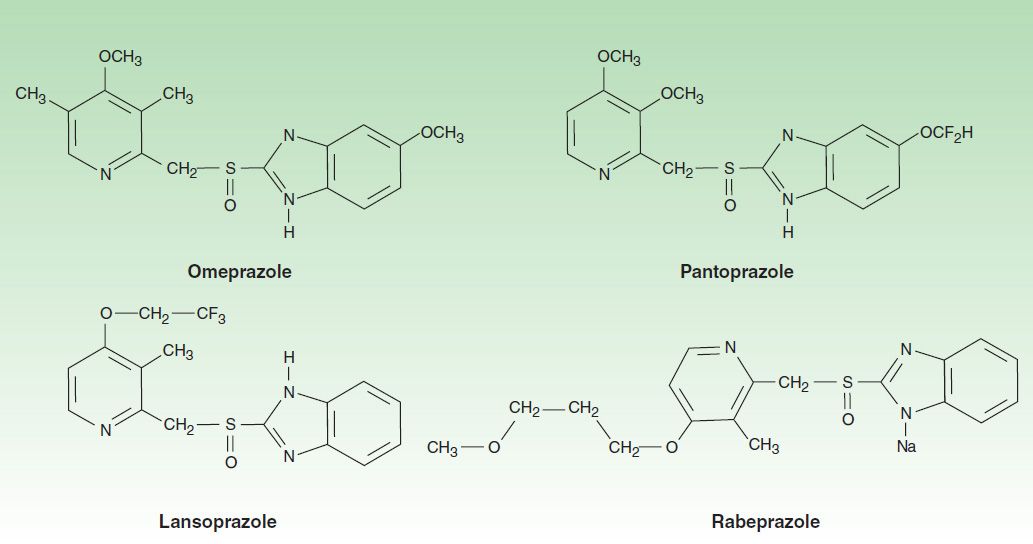
FIGURE 62–3 Molecular structure of the proton pump inhibitors: omeprazole, lansoprazole, pantoprazole, and the sodium salt of rabeprazole. Omeprazole and esomeprazole have the same chemical structure (see text).
TABLE 62–2 Pharmacokinetics of proton pump inhibitors.
PPIs are administered as inactive prodrugs. To protect the acid-labile prodrug from rapid destruction within the gastric lumen, oral products are formulated for delayed release as acid-resistant, enteric-coated capsules or tablets. After passing through the stomach into the alkaline intestinal lumen, the enteric coatings dissolve and the prodrug is absorbed. For children or patients with dysphagia or enteral feeding tubes, capsule formulations (but not tablets) may be opened and the microgranules mixed with apple or orange juice or mixed with soft foods (eg, applesauce). Esomeprazole, omeprazole, and pantoprazole are also available as oral suspensions. Lansoprazole is available as a tablet formulation that disintegrates in the mouth, and rabeprazole is available in a formulation that may be sprinkled on food. Omeprazole is also available as a powder formulation (capsule or packet) that contains sodium bicarbonate (1100–1680 mg NaHCO3; 304–460 mg of sodium) to protect the naked (non-enteric-coated) drug from acid degradation. When administered on an empty stomach by mouth or enteral tube, this “immediate-release” suspension results in rapid omeprazole absorption (Tmax < 30 minutes) and onset of acid inhibition.
The PPIs are lipophilic weak bases (pKa 4–5) and after intestinal absorption diffuse readily across lipid membranes into acidified compartments (eg, the parietal cell canaliculus). The prodrug rapidly becomes protonated within the canaliculus and is concentrated more than 1000-fold by Henderson-Hasselbalch trapping (see Chapter 1). There, it rapidly undergoes a molecular conversion to the active form, a reactive thiophilic sulfenamide cation, which forms a covalent disulfide bond with the H+/K+-ATPase, irreversibly inactivating the enzyme.
The pharmacokinetics of available PPIs are shown in Table 62–2. Immediate-release omeprazole has a faster onset of acid inhibition than other oral formulations. Although differences in pharmacokinetic profiles may affect speed of onset and duration of acid inhibition in the first few days of therapy, they are of little clinical importance with continued daily administration. The bioavailability of all agents is decreased approximately 50% by food; hence, the drugs should be administered on an empty stomach. In a fasting state, only 10% of proton pumps are actively secreting acid and susceptible to inhibition. PPIs should be administered approximately 1 hour before a meal (usually breakfast), so that the peak serum concentration coincides with the maximal activity of proton-pump secretion. The drugs have a short serum half-life of about 1.5 hours, but acid inhibition lasts up to 24 hours owing to the irreversible inactivation of the proton pump. At least 18 hours are required for synthesis of new H+/K+-ATPase pump molecules. Because not all proton pumps are inactivated with the first dose of medication, up to 3–4 days of daily medication are required before the full acid-inhibiting potential is reached. Similarly, after stopping the drug, it takes 3–4 days for full acid secretion to return.
PPIs undergo rapid first-pass and systemic hepatic metabolism and have negligible renal clearance. Dose reduction is not needed for patients with renal insufficiency or mild to moderate liver disease but should be considered in patients with severe liver impairment. Although other proton pumps exist in the body, the H+/K+-ATPase appears to exist only in the parietal cell and is distinct structurally and functionally from other H+-transporting enzymes.
The intravenous formulations of esomeprazole and pantoprazole have characteristics similar to those of the oral drugs. When given to a fasting patient, they inactivate acid pumps that are actively secreting, but they have no effect on pumps in quiescent, nonsecreting vesicles. Because the half-life of a single injection of the intravenous formulation is short, acid secretion returns several hours later as pumps move from the tubulovesicles to the canalicular surface. Thus, to provide maximal inhibition during the first 24–48 hours of treatment, the intravenous formulations must be given as a continuous infusion or as repeated bolus injections. The optimal dosing of intravenous PPIs to achieve maximal blockade in fasting patients is not yet established.
From a pharmacokinetic perspective, PPIs are ideal drugs: they have a short serum half-life, they are concentrated and activated near their site of action, and they have a long duration of action.
Pharmacodynamics
In contrast to H2 antagonists, PPIs inhibit both fasting and meal-stimulated secretion because they block the final common pathway of acid secretion, the proton pump. In standard doses, PPIs inhibit 90–98% of 24-hour acid secretion (Figure 62–2). When administered at equivalent doses, the different agents show little difference in clinical efficacy. In a crossover study of patients receiving long-term therapy with five PPIs, the mean 24-hour intragastric pH varied from 3.3 (pantoprazole, 40 mg) to 4.0 (esomeprazole, 40 mg) and the mean number of hours the pH was higher than 4 varied from 10.1 (pantoprazole, 40 mg) to 14.0 (esomeprazole, 40 mg). Although dexlansoprazole has a delayed-release formulation that results in a longer Tmax and greater AUC than other PPIs, it appears comparable to other agents in the ability to suppress acid secretion. This is because acid suppression is more dependent upon irreversible inactivation of the proton pump than the pharmacokinetics of different agents.
Clinical Uses
1. Gastroesophageal reflux disease—PPIs are the most effective agents for the treatment of nonerosive and erosive reflux disease, esophageal complications of reflux disease (peptic stricture or Barrett’s esophagus), and extraesophageal manifestations of reflux disease. Once-daily dosing provides effective symptom relief and tissue healing in 85–90% of patients; up to 15% of patients require twice-daily dosing.
GERD symptoms recur in over 80% of patients within 6 months after discontinuation of a PPI. For patients with erosive esophagitis or esophageal complications, long-term daily maintenance therapy with a full-dose or half-dose PPI is usually needed. Many patients with nonerosive GERD may be treated successfully with intermittent courses of PPIs or H2 antagonists taken as needed (“on demand”) for recurrent symptoms.
In current clinical practice, many patients with symptomatic GERD are treated empirically with medications without prior endoscopy, ie, without knowledge of whether the patient has erosive or nonerosive reflux disease. Empiric treatment with PPIs provides sustained symptomatic relief in 70–80% of patients, compared with 50–60% with H2 antagonists. Because of recent cost reductions, PPIs are used increasingly as first-line therapy for patients with symptomatic GERD.
Sustained acid suppression with twice-daily PPIs for at least 3 months is used to treat extraesophageal complications of reflux disease (asthma, chronic cough, laryngitis, and noncardiac chest pain).
2. Peptic ulcer disease—Compared with H2 antagonists, PPIs afford more rapid symptom relief and faster ulcer healing for duodenal ulcers and, to a lesser extent, gastric ulcers. All the pump inhibitors heal more than 90% of duodenal ulcers within 4 weeks and a similar percentage of gastric ulcers within 6–8 weeks.
a. H pylori-associated ulcers—For H pylori-associated ulcers, there are two therapeutic goals: to heal the ulcer and to eradicate the organism. The most effective regimens for H pylori eradication are combinations of two antibiotics and a PPI. PPIs promote eradication of H pylori through several mechanisms: direct antimicrobial properties (minor) and—by raising intragastric pH—lowering the minimal inhibitory concentrations of antibiotics against H pylori. The best treatment regimen consists of a 14-day regimen of “triple therapy”: a PPI twice daily; clarithromycin, 500 mg twice daily; and either amoxicillin, 1 g twice daily, or metronidazole, 500 mg twice daily. After completion of triple therapy, the PPI should be continued once daily for a total of 4–6 weeks to ensure complete ulcer healing. Alternatively, 10 days of “sequential treatment” consisting on days 1–5 of a PPI twice daily plus amoxicillin, 1 g twice daily, and followed on days 6–10 by five additional days of a PPI twice daily, plus clarithromycin, 500 mg twice daily, and tinidazole, 500 mg twice daily, has been shown to be a highly effective treatment regimen.
b. NSAID-associated ulcers—For patients with ulcers caused by aspirin or other NSAIDs, either H2 antagonists or PPIs provide rapid ulcer healing so long as the NSAID is discontinued; however continued use of the NSAID impairs ulcer healing. In patients with NSAID-induced ulcers who require continued NSAID therapy, treatment with a once- or twice-daily PPI more reliably promotes ulcer healing.
Asymptomatic peptic ulceration develops in 10–20% of people taking frequent NSAIDs, and ulcer-related complications (bleeding, perforation) develop in 1–2% of persons per year. PPIs taken once daily are effective in reducing the incidence of ulcers and ulcer complications in patients taking aspirin or other NSAIDs.
c. Prevention of rebleeding from peptic ulcers—In patients with acute gastrointestinal bleeding due to peptic ulcers, the risk of rebleeding from ulcers that have a visible vessel or adherent clot is increased. Rebleeding of this subset of high-risk ulcers is reduced significantly with PPIs administered for 3–5 days either as high-dose oral therapy (eg, omeprazole, 40 mg orally twice daily) or as a continuous intravenous infusion. It is believed that an intragastric pH higher than 6 may enhance coagulation and platelet aggregation. The optimal dose of intravenous PPI needed to achieve and maintain this level of near-complete acid inhibition is unknown; however, initial bolus administration of esomeprazole or pantoprazole (80 mg) followed by constant infusion (8 mg/h) is commonly recommended.
3. Nonulcer dyspepsia—PPIs have modest efficacy for treatment of nonulcer dyspepsia, benefiting 10–20% more patients than placebo. Despite their use for this indication, superiority to H2 antagonists (or even placebo) has not been conclusively demonstrated.
4. Prevention of stress-related mucosal bleeding—As discussed previously (see H2-Receptor Antagonists) PPIs (given orally, by nasogastric tube, or by intravenous infusions) may be administered to reduce the risk of clinically significant stress-related mucosal bleeding in critically ill patients. The only PPI approved by the FDA for this indication is an oral immediate-release omeprazole formulation, which is administered by nasogastric tube twice daily on the first day, then once daily. Although not FDA approved for this indication, other PPI suspension formulations (esomeprazole, omeprazole, pantoprazole) may also be used. For patients with nasoenteric tubes, PPI suspensions may be preferred to intravenous H2 antagonists or PPIs because of comparable efficacy, lower cost, and ease of administration.
For patients without a nasoenteric tube or with significant ileus, intravenous H2 antagonists are preferred to intravenous PPIs because of their proven efficacy. Although PPIs are increasingly used, there are no controlled trials demonstrating efficacy or optimal dosing.
5. Gastrinoma and other hypersecretory conditions—Patients with isolated gastrinomas are best treated with surgical resection. In patients with metastatic or unresectable gastrinomas, massive acid hypersecretion results in peptic ulceration, erosive esophagitis, and malabsorption. Previously, these patients required vagotomy and extraordinarily high doses of H2 antagonists, which still resulted in suboptimal acid suppression. With PPIs, excellent acid suppression can be achieved in all patients. Dosage is titrated to reduce basal acid output to less than 5–10 mEq/h. Typical doses of omeprazole are 60–120 mg/d.
Adverse Effects
1. General—PPIs are extremely safe. Diarrhea, headache, and abdominal pain are reported in 1–5% of patients, although the frequency of these events is only slightly increased compared with placebo. Increasing cases of acute interstitial nephritis have been reported. PPIs are not teratogenic in animal models; however, safety during pregnancy has not been established.
2. Nutrition—Acid is important in releasing vitamin B12 from food. A minor reduction in oral cyanocobalamin absorption occurs during proton pump inhibition, potentially leading to subnormal B12 levels with prolonged therapy. Acid also promotes absorption of food-bound minerals (non-heme iron, insoluble calcium salts, magnesium). Several case-control studies have suggested a modest increase in the risk of hip fracture in patients taking PPIs over a long term compared with matched controls. Although a causal relationship is unproven, PPIs may reduce calcium absorption or inhibit osteoclast function. Pending further studies, patients who require long-term PPIs—especially those with risk factors for osteoporosis—should have monitoring of bone density and should be provided calcium supplements. Cases of severe, life-threatening hypomagnesemia with secondary hypocalcemia due to PPIs have been reported; however, the mechanism of action is unknown.
3. Respiratory and enteric infections—Gastric acid is an important barrier to colonization and infection of the stomach and intestine from ingested bacteria. Increases in gastric bacterial concentrations are detected in patients taking PPIs, which is of unknown clinical significance. Some studies have reported an increased risk of both community-acquired respiratory infections and nosocomial pneumonia among patients taking PPIs.
There is a two- to threefold increased risk for hospital- and community-acquired Clostridium difficile infection in patients taking PPIs. There also is a small increased risk of other enteric infections (eg, Salmonella, Shigella, Escherichia coli, Campylobacter), which should be considered particularly when traveling in underdeveloped countries.
4. Potential problems due to increased serum gastrin—Gastrin levels are regulated by intragastric acidity. Acid suppression alters normal feedback inhibition so that median serum gastrin levels rise 1.5- to twofold in patients taking PPIs. Although gastrin levels remain within normal limits in most patients, they exceed 500 pg/mL (normal, < 100 pg/mL) in 3%. Upon stopping the drug, the levels normalize within 4 weeks. The rise in serum gastrin levels may stimulate hyperplasia of ECL and parietal cells, which may cause transient rebound acid hypersecretion with increased dyspepsia or heartburn after drug discontinuation, which abate within 2–4 weeks after gastrin and acid secretion normalize. In female rats given PPIs for prolonged periods, hypergastrinemia caused gastric carcinoid tumors that developed in areas of ECL hyperplasia. Although humans who take PPIs for a long time also may exhibit ECL hyperplasia, carcinoid tumor formation has not been documented. At present, routine monitoring of serum gastrin levels is not recommended in patients receiving prolonged PPI therapy.
5. Other potential problems due to decreased gastric acidity—Among patients infected with H pylori, long-term acid suppression leads to increased chronic inflammation in the gastric body and decreased inflammation in the antrum. Concerns have been raised that increased gastric inflammation may accelerate gastric gland atrophy (atrophic gastritis) and intestinal metaplasia—known risk factors for gastric adenocarcinoma. A special FDA Gastrointestinal Advisory Committee concluded that there is no evidence that prolonged PPI therapy produces the kind of atrophic gastritis (multifocal atrophic gastritis) or intestinal metaplasia that is associated with increased risk of adenocarcinoma. Routine testing for H pylori is not recommended in patients who require long-term PPI therapy. Long-term PPI therapy is associated with the development of small benign gastric fundic-gland polyps in a small number of patients, which may disappear after stopping the drug and are of uncertain clinical significance.
Drug Interactions
Decreased gastric acidity may alter absorption of drugs for which intragastric acidity affects drug bioavailability, eg, ketoconazole, itraconazole, digoxin, and atazanavir. All PPIs are metabolized by hepatic P450 cytochromes, including CYP2C19 and CYP3A4. Because of the short half-lives of PPIs, clinically significant drug interactions are rare. Omeprazole may inhibit the metabolism of warfarin, diazepam, and phenytoin. Esomeprazole also may decrease metabolism of diazepam. Lansoprazole may enhance clearance of theophylline. Rabeprazole and pantoprazole have no significant drug interactions.
The FDA has issued a warning about a potentially important adverse interaction between clopidogrel and PPIs. Clopidogrel is a prodrug that requires activation by the hepatic P450 CYP2C19 isoenzyme, which also is involved to varying degrees in the metabolism of PPIs (especially omeprazole, esomeprazole, lansoprazole, and dexlansoprazole). Thus, PPIs could reduce clopidogrel activation (and its antiplatelet action) in some patients. Several large retrospective studies have reported an increased incidence of serious cardiovascular events in patients taking clopidogrel and a PPI. In contrast, three smaller prospective randomized trials have not detected an increased risk. Pending further studies, PPIs should be prescribed to patients taking clopidogrel only if they have an increased risk of gastrointestinal bleeding or require them for chronic gastro-esophageal reflux or peptic ulcer disease, in which case agents with minimal CYP2C19 inhibition (pantoprazole or rabeprazole) are preferred.
MUCOSAL PROTECTIVE AGENTS
The gastroduodenal mucosa has evolved a number of defense mechanisms to protect itself against the noxious effects of acid and pepsin. Both mucus and epithelial cell-cell tight junctions restrict back diffusion of acid and pepsin. Epithelial bicarbonate secretion establishes a pH gradient within the mucous layer in which the pH ranges from 7 at the mucosal surface to 1–2 in the gastric lumen. Blood flow carries bicarbonate and vital nutrients to surface cells. Areas of injured epithelium are quickly repaired by restitution, a process in which migration of cells from gland neck cells seals small erosions to reestablish intact epithelium. Mucosal prostaglandins appear to be important in stimulating mucus and bicarbonate secretion and mucosal blood flow. A number of agents that potentiate these mucosal defense mechanisms are available for the prevention and treatment of acid-peptic disorders.
SUCRALFATE
Chemistry & Pharmacokinetics
Sucralfate is a salt of sucrose complexed to sulfated aluminum hydroxide. In water or acidic solutions it forms a viscous, tenacious paste that binds selectively to ulcers or erosions for up to 6 hours. Sucralfate has limited solubility, breaking down into sucrose sulfate (strongly negatively charged) and an aluminum salt. Less than 3% of intact drug and aluminum is absorbed from the intestinal tract; the remainder is excreted in the feces.
Pharmacodynamics
A variety of beneficial effects have been attributed to sucralfate, but the precise mechanism of action is unclear. It is believed that the negatively charged sucrose sulfate binds to positively charged proteins in the base of ulcers or erosion, forming a physical barrier that restricts further caustic damage and stimulates mucosal prostaglandin and bicarbonate secretion.
Clinical Uses
Sucralfate is administered in a dosage of 1 g four times daily on an empty stomach (at least 1 hour before meals). At present, its clinical uses are limited. Sucralfate (administered as a slurry through a nasogastric tube) reduces the incidence of clinically significant upper gastrointestinal bleeding in critically ill patients hospitalized in the intensive care unit, although it is slightly less effective than intravenous H2 antagonists. Sucralfate is still used by many clinicians for prevention of stress-related bleeding because of concerns that acid inhibitory therapies (antacids, H2 antagonists, and PPIs) may increase the risk of nosocomial pneumonia.
Adverse Effects
Because it is not absorbed, sucralfate is virtually devoid of systemic adverse effects. Constipation occurs in 2% of patients due to the aluminum salt. Because a small amount of aluminum is absorbed, it should not be used for prolonged periods in patients with renal insufficiency.
Drug Interactions
Sucralfate may bind to other medications, impairing their absorption.
PROSTAGLANDIN ANALOGS
Chemistry & Pharmacokinetics
The human gastrointestinal mucosa synthesizes a number of prostaglandins (see Chapter 18); the primary ones are prostaglandins E and F. Misoprostol, a methyl analog of PGE1, has been approved for gastrointestinal conditions. After oral administration, it is rapidly absorbed and metabolized to a metabolically active free acid. The serum half-life is less than 30 minutes; hence, it must be administered 3–4 times daily. It is excreted in the urine; however, dose reduction is not needed in patients with renal insufficiency.
Misoprostol has both acid inhibitory and mucosal protective properties. It is believed to stimulate mucus and bicarbonate secretion and enhance mucosal blood flow. Misoprostol can reduce the incidence of NSAID-induced ulcers to less than 3% and the incidence of ulcer complications by 50%. It is approved for prevention of NSAID-induced ulcers in high-risk patients; however, misoprostol has never achieved widespread use owing to its high adverse-effect profile and need for multiple daily dosing.
BISMUTH COMPOUNDS
Chemistry & Pharmacokinetics
Two bismuth compounds are available: bismuth subsalicylate, a nonprescription formulation containing bismuth and salicylate, and bismuth subcitrate potassium. In the USA, bismuth subcitrate is available only as a combination prescription product that also contains metronidazole and tetracycline for the treatment of H pylori. Bismuth subsalicylate undergoes rapid dissociation within the stomach, allowing absorption of salicylate. Over 99% of the bismuth appears in the stool. Although minimal (< 1%), bismuth is absorbed; it is stored in many tissues and has slow renal excretion. Salicylate (like aspirin) is readily absorbed and excreted in the urine.
Pharmacodynamics
The precise mechanisms of action of bismuth are unknown. Bismuth coats ulcers and erosions, creating a protective layer against acid and pepsin. It may also stimulate prostaglandin, mucus, and bicarbonate secretion. Bismuth subsalicylate reduces stool frequency and liquidity in acute infectious diarrhea, due to salicylate inhibition of intestinal prostaglandin and chloride secretion. Bismuth has direct antimicrobial effects and binds enterotoxins, accounting for its benefit in preventing and treating traveler’s diarrhea. Bismuth compounds have direct antimicrobial activity against H pylori.
Clinical Uses
In spite of the lack of comparative trials, nonprescription bismuth compounds (eg, Pepto-Bismol, Kaopectate) are widely used by patients for the nonspecific treatment of dyspepsia and acute diarrhea. Bismuth subsalicylate also is used for the prevention of traveler’s diarrhea (30 mL or 2 tablets four times daily).
Bismuth compounds are used in 4-drug regimens for the eradication of H pylori infection. One regimen consists of a PPI twice daily combined with bismuth subsalicylate (2 tablets; 262 mg each), tetracycline (250–500 mg), and metronidazole (500 mg) four times daily for 10–14 days. Another regimen consists of a PPI twice daily combined with three capsules of a combination prescription formulation (each capsule containing bismuth subcitrate 140 mg, metronidazole 125 mg, and tetracycline 125 mg) taken four times daily for 10 days. Although these are effective, standard “triple therapy” regimens (ie, PPI, clarithromycin, and amoxicillin or metronidazole twice daily for 14 days) generally are preferred for first-line therapy because of twice-daily dosing and superior compliance. Bismuth-based quadruple therapies are commonly used as second-line therapies.
Adverse Effects
All bismuth formulations have excellent safety profiles. Bismuth causes harmless blackening of the stool, which may be confused with gastrointestinal bleeding. Liquid formulations may cause harmless darkening of the tongue. Bismuth agents should be used for short periods only and should be avoided in patients with renal insufficiency. Prolonged usage of some bismuth compounds may rarely lead to bismuth toxicity, resulting in encephalopathy (ataxia, headaches, confusion, seizures). However, such toxicity is not reported with bismuth subsalicylate or bismuth citrate. High dosages of bismuth subsalicylate may lead to salicylate toxicity.
 DRUGS STIMULATING GASTROINTESTINAL MOTILITY
DRUGS STIMULATING GASTROINTESTINAL MOTILITY
Drugs that can selectively stimulate gut motor function (prokinetic agents) have significant potential clinical usefulness. Agents that increase lower esophageal sphincter pressures may be useful for GERD. Drugs that improve gastric emptying may be helpful for gastroparesis and postsurgical gastric emptying delay. Agents that stimulate the small intestine may be beneficial for postoperative ileus or chronic intestinal pseudo-obstruction. Finally, agents that enhance colonic transit may be useful in the treatment of constipation. Unfortunately, only a limited number of agents in this group are available for clinical use at this time.
PHYSIOLOGY OF THE ENTERIC NERVOUS SYSTEM
The enteric nervous system (see also Chapter 6) is composed of interconnected networks of ganglion cells and nerve fibers mainly located in the submucosa (submucosal plexus) and between the circular and longitudinal muscle layers (myenteric plexus). These networks give rise to nerve fibers that connect with the mucosa and muscle. Although extrinsic sympathetic and parasympathetic nerves project onto the submucosal and myenteric plexuses, the enteric nervous system can independently regulate gastrointestinal motility and secretion. Extrinsic primary afferent neurons project via the dorsal root ganglia or vagus nerve to the central nervous system (Figure 62–4). Release of serotonin (5-HT) from intestinal mucosa enterochromaffin (EC) cells stimulates 5-HT3 receptors on the extrinsic afferent nerves, stimulating nausea, vomiting, or abdominal pain. Serotonin also stimulates submucosal 5-HT1P receptors of the intrinsic primary afferent nerves (IPANs), which contain calcitonin gene-related peptide (CGRP) and acetylcholine and project to myenteric plexus interneurons. 5-HT4 receptors on the presynaptic terminals of the IPANs appear to enhance release of CGRP or acetylcholine. The myenteric interneurons are important in controlling the peristaltic reflex, promoting release of excitatory mediators proximally and inhibitory mediators distally. Motilin may stimulate excitatory neurons or muscle cells directly. Dopamine acts as an inhibitory neurotransmitter in the gastrointestinal tract, decreasing the intensity of esophageal and gastric contractions.
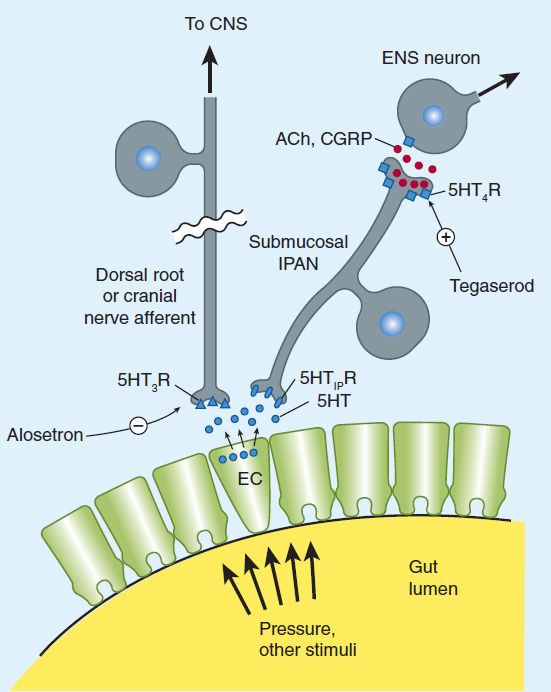
FIGURE 62–4 Release of serotonin (5-HT) by enterochromaffin (EC) cells from gut distention stimulates submucosal intrinsic primary afferent neurons (IPANs) via 5-HT1P receptors and extrinsic primary afferent neurons via 5-HT3 receptors (5-HT1PR, 5-HT3R). Submucosal IPANs activate the enteric neurons responsible for peristaltic and secretory reflex activity. Stimulation of 5-HT4 receptors (5-HT4R) on presynaptic terminals of IPANs enhances release of acetylcholine (ACh) and calcitonin gene-related peptide (CGRP), promoting reflex activity. CNS, central nervous system; ENS, enteric nervous system. (Data from Gershon MD: Serotonin and its implication for the management of irritable bowel syndrome. Rev Gastroenterol Dis 2003;3[Suppl 2]:S25.)
Although there are at least 14 serotonin receptor subtypes, 5-HT drug development for gastrointestinal applications to date has focused on 5-HT3-receptor antagonists and 5-HT4-receptor agonists.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


