13
Drugs Used in Heart Failure
CASE STUDY
A 65-year-old man developed shortness of breath with exertion several weeks after experiencing a viral illness. This was accompanied by swelling of the feet and ankles and increasing fatigue. On physical examination he is now found to be mildly short of breath lying down, but feels better sitting upright. Pulse is 105 bpm and regular, and blood pressure is 110/70 mm Hg. Crackles are noted at both lung bases, and his jugular venous pressure is elevated. The liver is enlarged, and there is 3+ edema of the ankles and feet. An echocardio-gram shows a dilated, poorly contracting heart with a left ventricular ejection fraction of about 20% (normal: 60%). The presumptive diagnosis is dilated cardiomyopathy secondary to a viral infection with stage C, class III heart failure. What treatment is indicated?
Heart failure occurs when cardiac output is inadequate to provide the oxygen needed by the body. It is a highly lethal condition, with a 5-year mortality rate conventionally said to be about 50%. The most common cause of heart failure in the USA is coronary artery disease, with hypertension also an important factor. Two major types of failure may be distinguished. Approximately 50% of younger patients have systolic failure, with reduced mechanical pumping action (contractility) and reduced ejection fraction. The remaining group has diastolic failure, with stiffening and loss of adequate relaxation playing a major role in reducing filling and cardiac output. Ejection fraction may be normal (preserved) in diastolic failure even though stroke volume is significantly reduced. The proportion of patients with diastolic failure increases with age. Because other cardiovascular conditions (especially myocardial infarction) are now being treated more effectively, more patients are surviving long enough for heart failure to develop, making heart failure one of the cardiovascular conditions that is actually increasing in prevalence.
Heart failure is a progressive disease that is characterized by a gradual reduction in cardiac performance, punctuated in many cases by episodes of acute decompensation, often requiring hospitalization. Treatment is therefore directed at two somewhat different goals: (1) reducing symptoms and slowing progression as much as possible during relatively stable periods and (2) managing acute episodes of decompensated failure. These factors are discussed in Clinical Pharmacology of Drugs Used in Heart Failure.
Although it is believed that the primary defect in early systolic heart failure resides in the excitation-contraction coupling machinery of the myocardium, the clinical condition also involves many other processes and organs, including the baroreceptor reflex, the sympathetic nervous system, the kidneys, angiotensin II and other peptides, aldosterone, and apoptosis of cardiac cells. Recognition of these factors has resulted in evolution of a variety of drug treatment strategies (Table 13–1).
TABLE 13–1 Therapies used in heart failure.
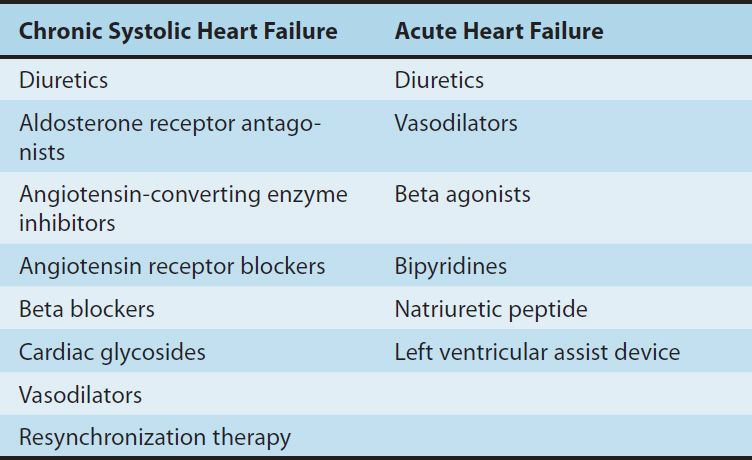
Large clinical trials have shown that therapy directed at noncardiac targets is more valuable in the long-term treatment of heart failure than traditional positive inotropic agents (cardiac glycosides [digitalis]). Extensive trials have shown that angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), certain β blockers, aldosterone receptor antagonists, and combined hydralazine-nitrate therapy are the only agents in current use that actually prolong life in patients with chronic heart failure. These strategies are useful in both systolic and diastolic failure. Positive inotropic drugs, on the other hand, are helpful mainly in acute systolic failure. Cardiac glycosides also reduce symptoms in chronic systolic heart failure. In large clinical trials to date, other positive inotropic drugs have usually reduced survival in chronic failure or had no benefit, and their use is discouraged.
Control of Normal Cardiac Contractility
The vigor of contraction of heart muscle is determined by several processes that lead to the movement of actin and myosin filaments in the cardiac sarcomere (Figure 13–1). Ultimately, contraction results from the interaction of activator calcium (during systole) with the actin-troponin-tropomyosin system, thereby releasing the actin-myosin interaction. This activator calcium is released from the sarcoplasmic reticulum (SR). The amount released depends on the amount stored in the SR and on the amount of trigger calcium that enters the cell during the plateau of the action potential.
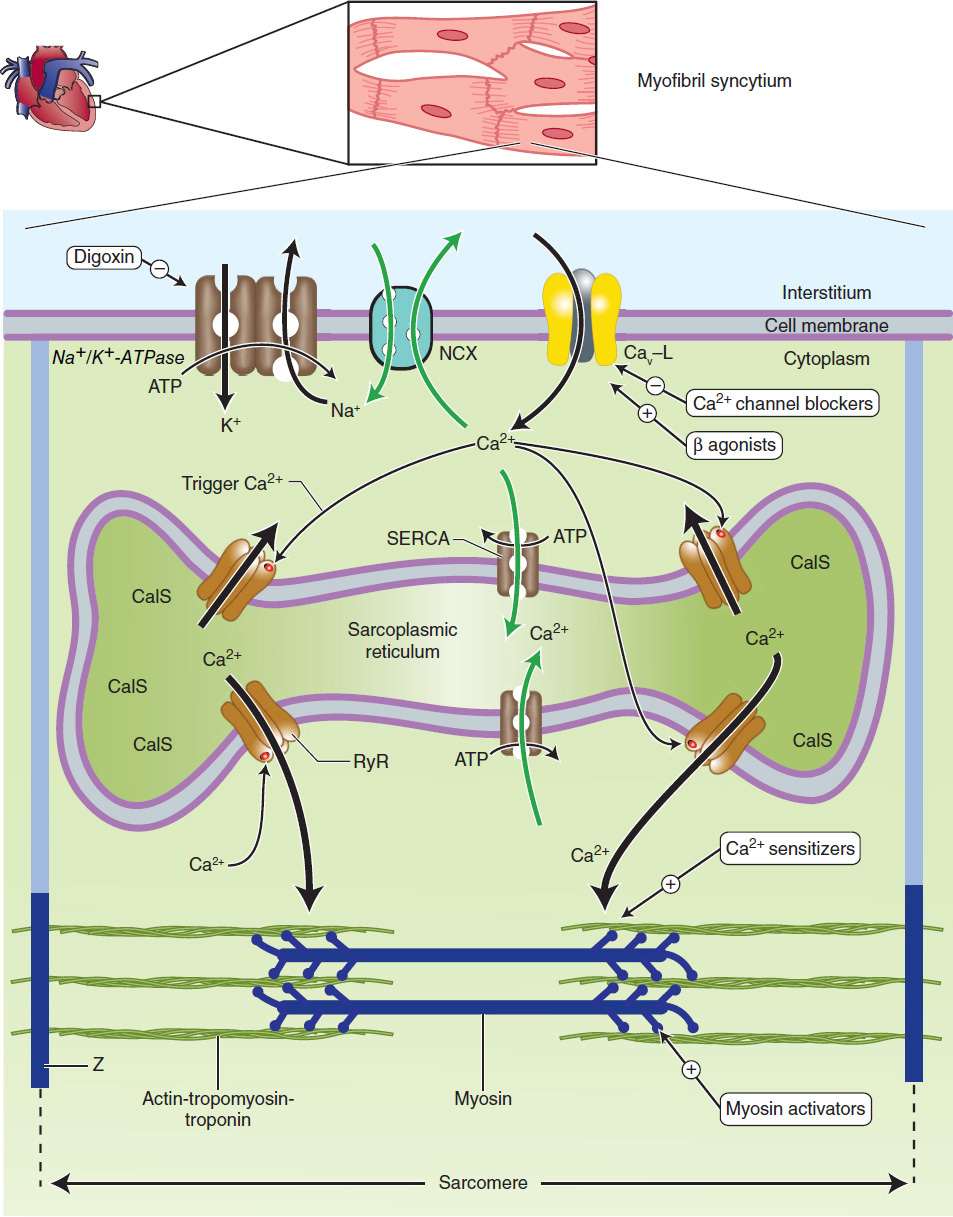
FIGURE 13–1 Schematic diagram of a cardiac muscle sarcomere, with sites of action of several drugs that alter contractility. Na+/K+-ATPase, the sodium pump, is the site of action of cardiac glycosides. NCX is the sodium-calcium exchanger. Cav-L is the voltage-gated, L-type calcium channel. SERCA (sarcoplasmic endoplasmic reticulum Ca2+-ATPase) is a calcium transporter ATPase that pumps calcium into the sarcoplasmic reticulum. CalS is calcium bound to calsequestrin, a high-capacity Ca2+-binding protein. RyR (ryanodine RyR2 receptor) is a calcium-activated calcium channel in the membrane of the SR that is triggered to release stored calcium. Z is the Z-line, which delimits the sarcomere. Calcium sensitizers act at the actin-troponin-tropomyosin complex where activator calcium brings about the contractile interaction of actin and myosin. Black arrows represent processes that initiate contraction or support basal tone. Green arrows represent processes that promote relaxation.
A. Sensitivity of the Contractile Proteins to Calcium and Other Contractile Protein Modifications
The determinants of calcium sensitivity, ie, the curve relating the shortening of cardiac myofibrils to the cytoplasmic calcium concentration, are incompletely understood, but several types of drugs can be shown to affect calcium sensitivity in vitro. Levosimendan is the most recent example of a drug that increases calcium sensitivity (it may also inhibit phosphodiesterase) and reduces symptoms in models of heart failure. A recent report suggests that an experimental drug, omecamtiv mecarbil(CK-1827452), alters the rate of transition of myosin from a low-actin-binding state to a strongly actin-bound, force-generating state. This action might increase contractility without increasing energy consumption, ie, increase efficiency.
B. Amount of Calcium Released from the Sarcoplasmic Reticulum
A small rise in free cytoplasmic calcium, brought about by calcium influx during the action potential, triggers the opening of calcium-gated, ryanodine-sensitive (RyR2) calcium channels in the membrane of the cardiac SR and the rapid release of a large amount of the ion into the cytoplasm in the vicinity of the actintroponin-tropomyosin complex. The amount released is proportional to the amount stored in the SR and the amount of trigger calcium that enters the cell through the cell membrane. (Ryanodine is a potent negative inotropic plant alkaloid that interferes with the release of calcium through cardiac SR channels.)
C. Amount of Calcium Stored in the Sarcoplasmic Reticulum
The SR membrane contains a very efficient calcium uptake transporter known as the sarcoplasmic endoplasmic reticulum Ca2+-ATPase (SERCA). This pump maintains free cytoplasmic calcium at very low levels during diastole by pumping calcium into the SR. SERCA is normally inhibited by phospholamban; phosphorylation of phospholamban by protein kinase A (activated, eg, by cAMP) removes this inhibition. The amount of calcium sequestered in the SR is thus determined, in part, by the amount accessible to this transporter and the activity of the sympathetic nervous system. This in turn is dependent on the balance of calcium influx (primarily through the voltage-gated membrane L-type calcium channels) and calcium efflux, the amount removed from the cell (primarily via the sodium-calcium exchanger, a transporter in the cell membrane). The amount of Ca2+ released from the SR depends on the response of the RyR channels to trigger Ca2+.
D. Amount of Trigger Calcium
The amount of trigger calcium that enters the cell depends on the concentration of extracellular calcium, the availability of membrane calcium channels, and the duration of their opening. As described in Chapters 6 and 9, sympathomimetics cause an increase in calcium influx through an action on these channels. Conversely, the calcium channel blockers (see Chapter 12) reduce this influx and depress contractility.
E. Activity of the Sodium-Calcium Exchanger
This antiporter (NCX) uses the sodium gradient to move calcium against its concentration gradient from the cytoplasm to the extracellular space. Extracellular concentrations of these ions are much less labile than intracellular concentrations under physiologic conditions. The sodium-calcium exchanger’s ability to carry out this transport is thus strongly dependent on the intracellular concentrations of both ions, especially sodium.
F. Intracellular Sodium Concentration and Activity of Na+/K+-ATPase
Na+/K+-ATPase, by removing intracellular sodium, is the major determinant of sodium concentration in the cell. The sodium influx through voltage-gated channels, which occurs as a normal part of almost all cardiac action potentials, is another determinant, although the amount of sodium that enters with each action potential is much less than 1% of the total intracellular sodium. Na+/K+-ATPase appears to be the primary target of digoxin and other cardiac glycosides.
Pathophysiology of Heart Failure
Heart failure is a syndrome with many causes that may involve one or both ventricles. Cardiac output is usually below the normal range (“low-output” failure). Systolic dysfunction, with reduced cardiac output and significantly reduced ejection fraction (EF < 45%; normal > 60%), is typical of acute failure, especially that resulting from myocardial infarction. Diastolic dysfunction often occurs as a result of hypertrophy and stiffening of the myocardium, and although cardiac output is reduced, ejection fraction may be normal. Heart failure due to diastolic dysfunction does not usually respond optimally to positive inotropic drugs.
“High-output” failure is a rare form of heart failure. In this condition, the demands of the body are so great that even increased cardiac output is insufficient. High-output failure can result from hyperthyroidism, beriberi, anemia, and arteriovenous shunts. This form of failure responds poorly to the drugs discussed in this chapter and should be treated by correcting the underlying cause.
The primary signs and symptoms of all types of heart failure include tachycardia, decreased exercise tolerance, shortness of breath, and cardiomegaly. Peripheral and pulmonary edema (the congestion of congestive heart failure) are often but not always present. Decreased exercise tolerance with rapid muscular fatigue is the major direct consequence of diminished cardiac output. The other manifestations result from the attempts by the body to compensate for the intrinsic cardiac defect.
Neurohumoral (extrinsic) compensation involves two major mechanisms (previously presented in Figure 6–7)—the sympathetic nervous system and the renin-angiotensin-aldosterone hormonal response—plus several others. Some of the detrimental as well as beneficial features of these compensatory responses are illustrated in Figure 13–2. The baroreceptor reflex appears to be reset, with a lower sensitivity to arterial pressure, in patients with heart failure. As a result, baroreceptor sensory input to the vasomotor center is reduced even at normal pressures; sympathetic outflow is increased, and parasympathetic outflow is decreased. Increased sympathetic outflow causes tachycardia, increased cardiac contractility, and increased vascular tone. Vascular tone is further increased by angiotensin II and endothelin, a potent vasoconstrictor released by vascular endothelial cells. Vasoconstriction increases afterload, which further reduces ejection fraction and cardiac output. The result is a vicious cycle that is characteristic of heart failure (Figure 13–3). Neurohumoral antagonists and vasodilators reduce heart failure mortality by interrupting the cycle and slowing the downward spiral.
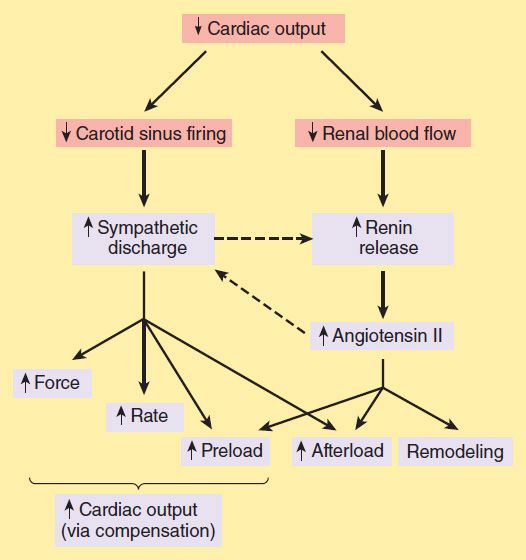
FIGURE 13–2 Some compensatory responses (orange boxes) that occur during congestive heart failure. In addition to the effects shown, sympathetic discharge facilitates renin release, and angiotensin II increases norepinephrine release by sympathetic nerve endings (dashed arrows).
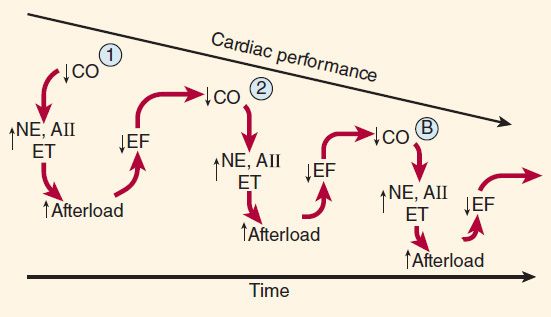
FIGURE 13–3 Vicious spiral of progression of heart failure. Decreased cardiac output (CO) activates production of neurohormones (NE, norepinephrine; AII, angiotensin II; ET, endothelin), which cause vasoconstriction and increased afterload. This further reduces ejection fraction (EF) and CO, and the cycle repeats. The downward spiral is continued until a new steady state is reached in which CO is lower and afterload is higher than is optimal for normal activity. Circled points 1, 2, and B represent points on the ventricular function curves depicted in Figure 13–4.
After a relatively short exposure to increased sympathetic drive, complex down-regulatory changes in the cardiac β1-adrenoceptor–G protein-effector system take place that result in diminished stimulatory effects. Beta2 receptors are not down-regulated and may develop increased coupling to the inositol 1,4,5-trisphosphate–diacylglycerol (IP3-DAG) cascade. It has also been suggested that cardiac β3 receptors (which do not appear to be down-regulated in failure) may mediate negative inotropic effects. Excessive β activation can lead to leakage of calcium from the SR via RyR channels and contributes to stiffening of the ventricles and arrhythmias. Prolonged β activation also increases caspases, the enzymes responsible for apoptosis. Increased angiotensin II production leads to increased aldosterone secretion (with sodium and water retention), to increased afterload, and to remodeling of both heart and vessels. Other hormones are released, including natriuretic peptide, endothelin, and vasopressin (see Chapter 17). Within the heart, failure-induced changes have been documented in calcium handling in the SR by SERCA and phospholamban; in transcription factors that lead to hypertrophy and fibrosis; in mitochondrial function, which is critical for energy production in the overworked heart; and in ion channels, especially potassium channels, which facilitate arrhythmo-genesis, a primary cause of death in heart failure. Phosphorylation of RyR channels in the sarcoplasmic reticulum enhances and dephosphorylation reduces Ca2+ release; studies in animal models indicate that the enzyme primarily responsible for RyR dephosphorylation, protein phosphatase 1 (PP1), is up-regulated in heart failure. These cellular changes provide many potential targets for future drugs.
The most important intrinsic compensatory mechanism is myocardial hypertrophy. The increase in muscle mass helps maintain cardiac performance. However, after an initial beneficial effect, hypertrophy can lead to ischemic changes, impairment of diastolic filling, and alterations in ventricular geometry. Remodeling is the term applied to dilation (other than that due to passive stretch) and other slow structural changes that occur in the stressed myocardium. It may include proliferation of connective tissue cells as well as abnormal myocardial cells with some biochemical characteristics of fetal myocytes. Ultimately, myocytes in the failing heart die at an accelerated rate through apoptosis, leaving the remaining myocytes subject to even greater stress.
Pathophysiology of Cardiac Performance
Cardiac performance is a function of four primary factors:
1. Preload: When some measure of left ventricular performance such as stroke volume or stroke work is plotted as a function of left ventricular filling pressure or end-diastolic fiber length, the resulting curve is termed the left ventricular function curve (Figure 13–4). The ascending limb (< 15 mm Hg filling pressure) represents the classic Frank-Starling relation described in physiology texts. Beyond approximately 15 mm Hg, there is a plateau of performance. Preloads greater than 20–25 mm Hg result in pulmonary congestion. As noted above, preload is usually increased in heart failure because of increased blood volume and venous tone. Because the function curve of the failing heart is lower, the plateau is reached at much lower values of stroke work or output. Increased fiber length or filling pressure increases oxygen demand in the myocardium, as described in Chapter 12. Reduction of high filling pressure is the goal of salt restriction and diuretic therapy in heart failure. Venodilator drugs (eg, nitroglycerin) also reduce preload by redistributing blood away from the chest into peripheral veins.
2. Afterload: Afterload is the resistance against which the heart must pump blood and is represented by aortic impedance and systemic vascular resistance. As noted in Figure 13–2, as cardiac output falls in chronic failure, a reflex increase in systemic vascular resistance occurs, mediated in part by increased sympathetic outflow and circulating catecholamines and in part by activation of the renin-angiotensin system. Endothelin, a potent vasoconstrictor peptide, is also involved. This sets the stage for the use of drugs that reduce arteriolar tone in heart failure.
3. Contractility: Heart muscle obtained by biopsy from patients with chronic low-output failure demonstrates a reduction in intrinsic contractility. As contractility decreases in the heart, there is a reduction in the velocity of muscle shortening, the rate of intraventricular pressure development (dP/dt), and the stroke output achieved (Figure 13–4). However, the heart is usually still capable of some increase in all of these measures of contractility in response to inotropic drugs.
4. Heart rate: The heart rate is a major determinant of cardiac output. As the intrinsic function of the heart decreases in failure and stroke volume diminishes, an increase in heart rate—through sympathetic activation of β adrenoceptors—is the first compensatory mechanism that comes into play to maintain cardiac output.
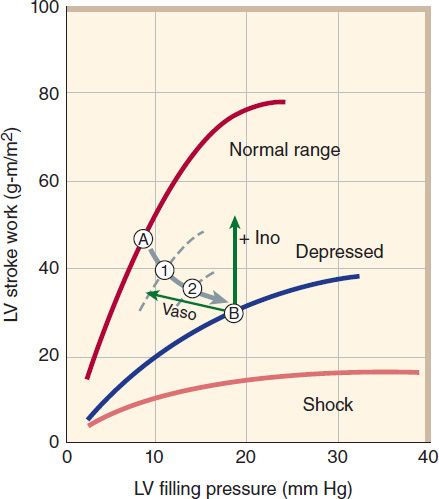
FIGURE 13–4 Relation of left ventricular (LV) performance to filling pressure in patients with acute myocardial infarction, an important cause of heart failure. The upper line indicates the range for normal, healthy individuals. At a given level of exercise, the heart operates at a stable point, eg, point A. In heart failure, function is shifted down and to the right, through points 1 and 2, finally reaching point B. A “pure” positive inotropic drug (+ Ino) would move the operating point upward by increasing cardiac stroke work. A vasodilator (Vaso) would move the point leftward by reducing filling pressure. Successful therapy usually results in both effects. (Adapted, with permission, from Swan HJC, Parmley WW: Congestive heart failure. In: Sodeman WA Jr, Sodeman TM [editors]: Pathologic Physiology, 7th ed. Saunders, 1985. Copyright Elsevier.)
 BASIC PHARMACOLOGY OF DRUGS USED IN HEART FAILURE
BASIC PHARMACOLOGY OF DRUGS USED IN HEART FAILURE
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


