34
Drugs Used in Disorders of Coagulation
CASE STUDY
A 25-year-old woman presents to the emergency department complaining of acute onset of shortness of breath and pleuritic pain. She had been in her usual state of health until 2 days prior when she noted that her left leg was swollen and red. Her only medication was oral contraceptives. Family history was significant for a history of “blood clots” in multiple members of the maternal side of her family. Physical examination demonstrates an anxious woman with stable vital signs. The left lower extremity demonstrates erythema and edema and is tender to touch. Ultrasound reveals a deep vein thrombosis in the left lower extremity; chest computed tomography scan confirms the presence of pulmonary emboli. Laboratory blood tests indicate elevated D-dimer levels. What therapy is indicated acutely? What are the long-term therapy options? How long should she be treated? Should this individual use oral contraceptives?
Hemostasis refers to the finely regulated dynamic process of maintaining fluidity of the blood, repairing vascular injury, and limiting blood loss while avoiding vessel occlusion (thrombosis) and inadequate perfusion of vital organs. Either extreme—excessive bleeding or thrombosis—represents a breakdown of the hemostatic mechanism. Common causes of dysregulated hemostasis include hereditary or acquired defects in the clotting mechanism and secondary effects of infection or cancer. The drugs used to inhibit thrombosis and to limit abnormal bleeding are the subjects of this chapter.
MECHANISMS OF BLOOD COAGULATION
The vascular endothelial cell layer lining blood vessels has an anticoagulant phenotype, and circulating blood platelets and clotting factors do not normally adhere to it to an appreciable extent. In the setting of vascular injury, the endothelial cell layer rapidly undergoes a series of changes resulting in a more procoagulant phenotype. Injury exposes reactive subendothelial matrix proteins such as collagen and von Willebrand factor, which results in platelet adherence and activation, and secretion and synthesis of vasoconstrictors and platelet-recruiting and activating molecules. Thus, thromboxane A2 (TXA2) is synthesized from arachidonic acid within platelets and is a platelet activator and potent vasoconstrictor. Products secreted from platelet granules include adenosine diphosphate (ADP), a powerful inducer of platelet aggregation, and serotonin (5-HT), which stimulates aggregation and vasoconstriction. Activation of platelets results in a conformational change in the αIIbβIII integrin (IIb/IIIa) receptor, enabling it to bind fibrinogen, which cross-links adjacent platelets, resulting in aggregation and formation of a platelet plug (Figure 34–1). Simultaneously, the coagulation system cascade is activated, resulting in thrombin generation and a fibrin clot, which stabilizes the platelet plug (see below). Knowledge of the hemostatic mechanism is important for diagnosis of bleeding disorders. Patients with defects in the formation of the primary platelet plug (defects in primary hemostasis, eg, platelet function defects, von Willebrand disease) typically bleed from surface sites (gingiva, skin, heavy menses) with injury. In contrast, patients with defects in the clotting mechanism (secondary hemostasis, eg, hemophilia A) tend to bleed into deep tissues (joints, muscle, retroperitoneum), often with no apparent inciting event, and bleeding may recur unpredictably.
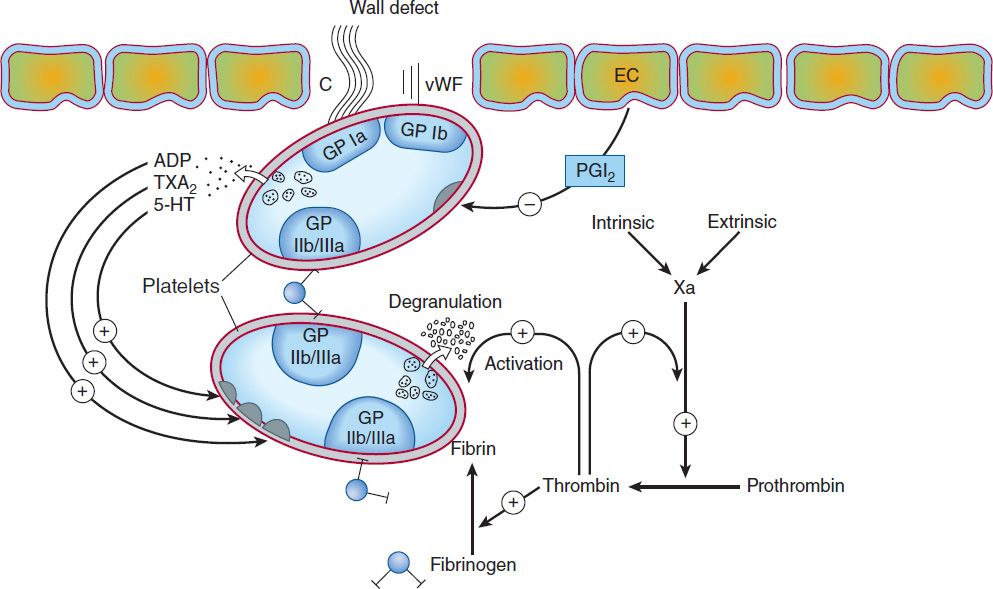
FIGURE 34–1 Thrombus formation at the site of the damaged vascular wall (EC, endothelial cell) and the role of platelets and clotting factors. Platelet membrane receptors include the glycoprotein (GP) Ia receptor, binding to collagen (C); GP Ib receptor, binding von Willebrand factor (vWF); and GP IIb/IIIa, which binds fibrinogen and other macromolecules. Antiplatelet prostacyclin (PGI2) is released from the endothelium. Aggregating substances released from the degranulating platelet include adenosine diphosphate (ADP), thromboxane A2 (TXA2), and serotonin (5-HT). Production of factor Xa by intrinsic and extrinsic pathways is detailed in Figure 34–2. (Redrawn and reproduced, with permission, from Simoons ML, Decker JW: New directions in anticoagulant and antiplatelet treatment. [Editorial.] Br Heart J 1995;74:337.)
The platelet is central to normal hemostasis and thromboembolic disease, and is the target of many therapies discussed in this chapter. Platelet-rich thrombi (white thrombi) form in the high flow rate and high shear force environment of arteries. Occlusive arterial thrombi cause serious disease by producing downstream ischemia of extremities or vital organs, and can result in limb amputation or organ failure. Venous clots tend to be more fibrin-rich, contain large numbers of trapped red blood cells, and are recognized pathologically as red thrombi. Deep venous thrombi (DVT) can cause severe swelling and pain of the affected extremity, but the most feared consequence is pulmonary embolism (PE). This occurs when part or all of the clot breaks off from its location in the deep venous system and travels as an embolus through the right side of the heart and into the pulmonary arterial circulation. Occlusion of a large pulmonary artery by an embolic clot can precipitate acute right heart failure and sudden death. In addition lung ischemia or infarction will occur distal to the occluded pulmonary arterial segment. Such emboli usually arise from the deep venous system of the proximal lower extremities or pelvis. Although all thrombi are mixed, the platelet nidus dominates the arterial thrombus and the fibrin tail dominates the venous thrombus.
BLOOD COAGULATION CASCADE
Blood coagulates due to the transformation of soluble fibrinogen into insoluble fibrin by the enzyme thrombin. Several circulating proteins interact in a cascading series of limited proteolytic reactions (Figure 34–2). At each step, a clotting factor zymogen undergoes limited proteolysis and becomes an active protease (eg, factor VII is converted to factor VIIa). Each protease factor activates the next clotting factor in the sequence, culminating in the formation of thrombin (factor IIa). Several of these factors are targets for drug therapy (Table 34–1).
TABLE 34–1 Blood clotting factors and drugs that affect them.1
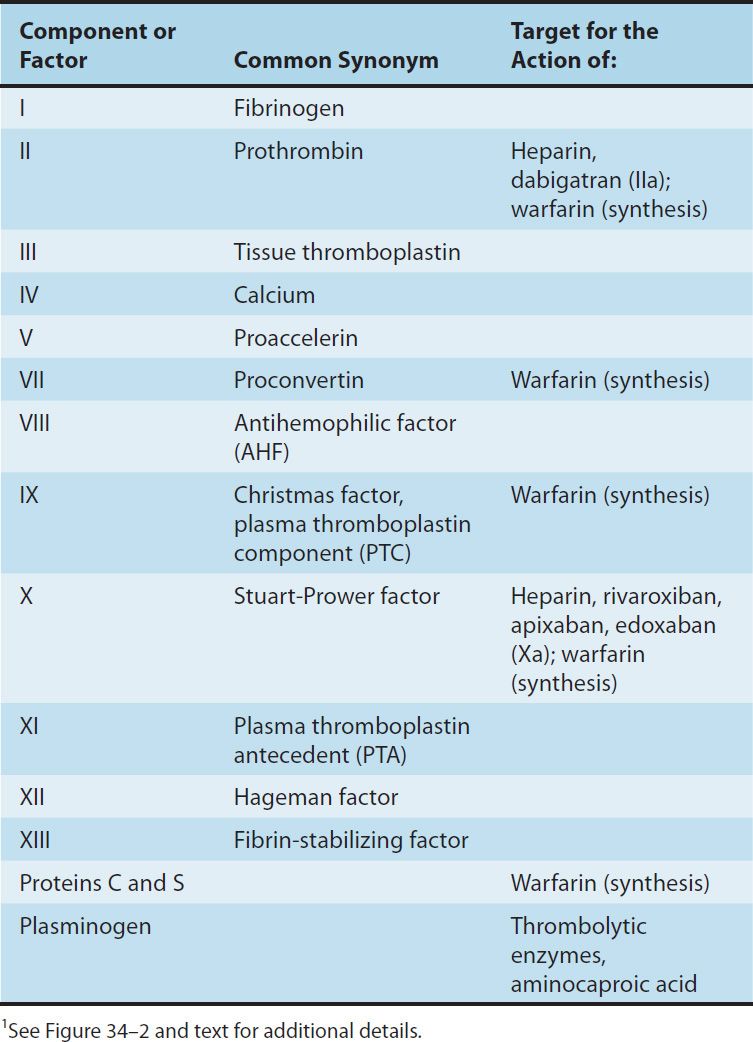
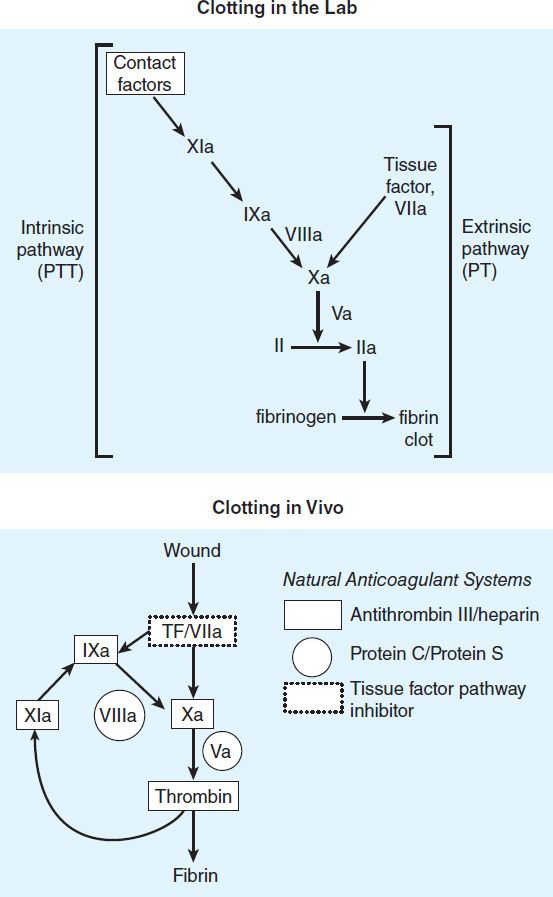
FIGURE 34–2 A model of blood coagulation. With tissue factor (TF), factor VII forms an activated complex (VIIa-TF) that catalyzes the activation of factor IX to factor IXa. Activated factor XIa also catalyzes this reaction. Tissue factor pathway inhibitor inhibits the catalytic action of the VIIa-TF complex. The cascade proceeds as shown, resulting ultimately in the conversion of fibrinogen to fibrin, an essential component of a functional clot. The two major anticoagulant drugs, heparin and warfarin, have very different actions. Heparin, acting in the blood, directly activates anticlotting factors, specifically antithrombin, which inactivates the factors enclosed in rectangles. Warfarin, acting in the liver, inhibits the synthesis of the factors enclosed in circles. Proteins C and S exert anticlotting effects by inactivating activated factors Va and VIIIa.
Thrombin has a central role in hemostasis and has many functions. In clotting, thrombin proteolytically cleaves small peptides from fibrinogen, allowing fibrinogen to polymerize and form a fibrin clot. Thrombin also activates many upstream clotting factors, leading to more thrombin generation, and activates factor XIII, a transaminase that cross-links the fibrin polymer and stabilizes the clot. Thrombin is a potent platelet activator and mitogen. Thrombin also exerts anticoagulant effects by activating the protein C pathway, which attenuates the clotting response (Figure 34–2). It should therefore be apparent that the response to vascular injury is a complex and precisely modulated process that ensures that under normal circumstances, repair of vascular injury occurs without thrombosis and downstream ischemia; that is, the response is proportionate and reversible. Eventually vascular remodeling and repair occur with reversion to the quiescent resting anticoagulant endothelial cell phenotype.
Initiation of Clotting: The Tissue Factor-VIIa Complex
The main initiator of blood coagulation in vivo is the tissue factor (TF)-factor VIIa pathway (Figure 34–2). Tissue factor is a transmembrane protein ubiquitously expressed outside the vasculature, but not normally expressed in an active form within vessels. The exposure of TF on damaged endothelium or to blood that has extravasated into tissue binds TF to factor VIIa. This complex, in turn, activates factors X and IX. Factor Xa along with factor Va forms the prothrombinase complex on activated cell surfaces, which catalyzes the conversion of prothrombin (factor II) to thrombin (factor IIa). Thrombin, in turn, activates upstream clotting factors, primarily factors V, VIII, and XI, resulting in amplification of thrombin generation. The TF-factor VIIa-catalyzed activation of factor Xa is regulated by tissue factor pathway inhibitor (TFPI). Thus after initial activation of factor X to Xa by TF-VIIa, further propagation of the clot is by feedback amplification of thrombin through the intrinsic pathway factors VIII and IX (this provides an explanation of why patients with deficiency of factor VIII or IX—hemophilia A and hemophilia B, respectively—have a severe bleeding disorder).
It is also important to note that the coagulation mechanism in vivo does not occur in solution, but is localized to activated cell surfaces expressing anionic phospholipids such as phosphatidylserine, and is mediated by Ca2+ bridging between the anionic phospholipids and γ-carboxyglutamic acid residues of the clotting factors. This is the basis for using calcium chelators such as ethylenediamine tetraacetic acid (EDTA) or citrate to prevent blood from clotting in a test tube.
Antithrombin (AT) is an endogenous anticoagulant and a member of the serine protease inhibitor (serpin) family; it inactivates the serine proteases IIa, IXa, Xa, XIa, and XIIa. The endogenous anticoagulants protein C and protein S attenuate the blood clotting cascade by proteolysis of the two cofactors Va and VIIIa. From an evolutionary standpoint, it is of interest that factors V and VIII have an identical overall domain structure and considerable homology, consistent with a common ancestor gene; likewise the serine proteases are descendants of a trypsin-like common ancestor. Thus, the TF-VIIa initiating complex, serine proteases, and cofactors each have their own lineage-specific attenuation mechanism (Figure 34–2). Defects in natural anticoagulants result in an increased risk of venous thrombosis. The most common defect in the natural anticoagulant system is a mutation in factor V (factor V Leiden), which results in resistance to inactivation by the protein C, protein S mechanism.
Fibrinolysis
Fibrinolysis refers to the process of fibrin digestion by the fibrin-specific protease, plasmin. The fibrinolytic system is similar to the coagulation system in that the precursor form of the serine protease plasmin circulates in an inactive form as plasminogen. In response to injury, endothelial cells synthesize and release tissue plasminogen activator (t-PA), which converts plasminogen to plasmin (Figure 34–3). Plasmin remodels the thrombus and limits its extension by proteolytic digestion of fibrin.
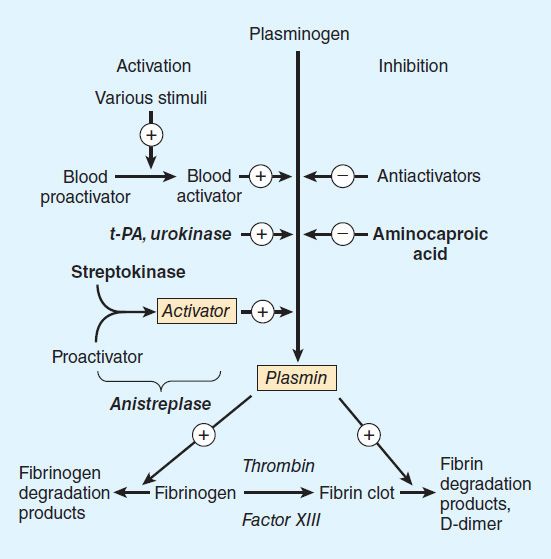
FIGURE 34–3 Schematic representation of the fibrinolytic system. Plasmin is the active fibrinolytic enzyme. Several clinically useful activators are shown on the left in bold. Anistreplase is a combination of streptokinase and the proactivator plasminogen. Aminocaproic acid (right) inhibits the activation of plasminogen to plasmin and is useful in some bleeding disorders. t-PA, tissue plasminogen activator.
Both plasminogen and plasmin have specialized protein domains (kringles) that bind to exposed lysines on the fibrin clot and impart clot specificity to the fibrinolytic process. It should be noted that this clot specificity is only observed at physiologic levels of t-PA. At the pharmacologic levels of t-PA used in thrombolytic therapy, clot specificity is lost and a systemic lytic state is created, with attendant increase in bleeding risk. As in the coagulation cascade, there are negative regulators of fibrinolysis: endothelial cells synthesize and release plasminogen activator inhibitor (PAI), which inhibits t-PA; in addition α2 antiplasmin circulates in the blood at high concentrations and under physiologic conditions will rapidly inactivate any plasmin that is not clot-bound. However, this regulatory system is overwhelmed by therapeutic doses of plasminogen activators.
If the coagulation and fibrinolytic systems are pathologically activated, the hemostatic system may careen out of control, leading to generalized intravascular clotting and bleeding. This process is called disseminated intravascular coagulation (DIC) and may follow massive tissue injury, advanced cancers, obstetric emergencies such as abruptio placentae or retained products of conception, or bacterial sepsis. The treatment of DIC is to control the underlying disease process; if this is not possible, DIC is often fatal.
Regulation of the fibrinolytic system is useful in therapeutics. Increased fibrinolysis is effective therapy for thrombotic disease. Tissue plasminogen activator, urokinase, and streptokinase all activate the fibrinolytic system (Figure 34–3). Conversely, decreased fibrinolysis protects clots from lysis and reduces the bleeding of hemostatic failure. Aminocaproic acid is a clinically useful inhibitor of fibrinolysis. Heparin and the oral anticoagulant drugs do not affect the fibrinolytic mechanism.
 BASIC PHARMACOLOGY OF THE ANTICOAGULANT DRUGS
BASIC PHARMACOLOGY OF THE ANTICOAGULANT DRUGS
The ideal anticoagulant drug would prevent pathologic thrombosis and limit reperfusion injury, yet allow a normal response to vascular injury and limit bleeding. Theoretically this could be accomplished by preservation of the TF-VIIa initiation phase of the clotting mechanism with attenuation of the secondary intrinsic pathway propagation phase of clot development. At this time such a drug does not exist; all anticoagulants and fibrinolytic drugs have an increased bleeding risk as their principle toxicity.
INDIRECT THROMBIN INHIBITORS
The indirect thrombin inhibitors are so-named because their antithrombotic effect is exerted by their interaction with a separate protein, antithrombin. Unfractionated heparin (UFH), also known as high-molecular-weight (HMW) heparin, low-molecular-weight (LMW) heparin, and the synthetic pentasaccharide fondaparinux bind to antithrombin and enhance its inactivation of factor Xa (Figure 34–4). Unfractionated heparin and to a lesser extent LMW heparin also enhance antithrombin’s inactivation of thrombin.
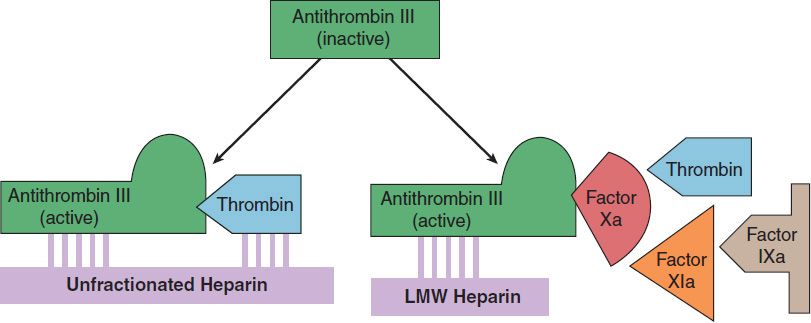
FIGURE 34–4 Cartoon illustrating differences between low-molecular-weight (LMW) heparins and high-molecular-weight heparin (unfractionated heparin). Fondaparinux is a small pentasaccharide fragment of heparin. Activated antithrombin III (AT III) degrades thrombin, factor X, and several other factors. Binding of these drugs to AT III can increase the catalytic action of AT III 1000-fold. The combination of AT III with unfractionated heparin increases degradation of both factor Xa and thrombin. Combination with fondaparinux or LMW heparin more selectively increases degradation of Xa.
HEPARIN
Chemistry & Mechanism of Action
Heparin is a heterogeneous mixture of sulfated mucopolysaccharides. It binds to endothelial cell surfaces and a variety of plasma proteins. Its biologic activity is dependent upon the endogenous anticoagulant antithrombin. Antithrombin inhibits clotting factor proteases, especially thrombin (IIa), IXa, and Xa, by forming equimolar stable complexes with them. In the absence of heparin, these reactions are slow; in the presence of heparin, they are accelerated 1000-fold. Only about a third of the molecules in commercial heparin preparations have an accelerating effect because the remainder lack the unique pentasaccharide sequence needed for high-affinity binding to antithrombin. The active heparin molecules bind tightly to antithrombin and cause a conformational change in this inhibitor. The conformational change of antithrombin exposes its active site for more rapid interaction with the proteases (the activated clotting factors). Heparin functions as a cofactor for the antithrombin-protease reaction without being consumed. Once the antithrombin-protease complex is formed, heparin is released intact for renewed binding to more antithrombin.
The antithrombin binding region of commercial unfractionated heparin consists of repeating sulfated disaccharide units composed of D-glucosamine-L-iduronic acid and D-glucosamine-D-glucuronic acid. High-molecular-weight fractions of heparin with high affinity for antithrombin markedly inhibit blood coagulation by inhibiting all three factors, especially thrombin and factor Xa. Unfractionated heparin has a molecular weight range of 5000–30,000. In contrast, the shorter-chain, low-molecular-weight fractions of heparin inhibit activated factor X but have less effect on thrombin than the HMW species. Nevertheless, numerous studies have demonstrated that LMW heparins such as enoxaparin, dalteparin, and tinzaparin are effective in several thromboembolic conditions. In fact, these LMW heparins—in comparison with UFH—have equal efficacy, increased bioavailability from the subcutaneous site of injection, and less frequent dosing requirements (once or twice daily is sufficient).
Because commercial heparin consists of a family of molecules of different molecular weights extracted from porcine intestinal mucosa and bovine lung, the correlation between the concentration of a given heparin preparation and its effect on coagulation often is poor. Therefore, UFH is standardized by bioassay. Heparin was reformulated in 2009 in response to heparin contamination events in 2007 and 2008. The contaminant was identified as over-sulfated chondroitin sulfate and linked to more than150 adverse events in patients, most commonly hypotension, nausea, and dyspnea within 30 minutes of infusion. In response to this event, heparin sodium was reformulated with stricter quality control measures and bioassays to make detection of contaminants easier. This reformulation led to a decrease in potency of approximately 10% from the previous formulation. USP heparin is now harmonized to the World Health Organization International Standard (IS) unit dose. Enoxaparin is obtained from the same sources as regular UFH, but doses are specified in milligrams. Fondaparinux is also specified in milligrams. Dalteparin, tinzaparin, and danaparoid (an LMW heparinoid containing heparan sulfate, dermatan sulfate, and chondroitin sulfate), on the other hand, are specified in anti-factor Xa units.
Monitoring of Heparin Effect
Close monitoring of the activated partial thromboplastin time (aPTT or PTT) is necessary in patients receiving UFH. Levels of UFH may also be determined by protamine titration (therapeutic levels 0.2–0.4 unit/mL) or anti-Xa units (therapeutic levels 0.3–0.7 unit/mL). Weight-based dosing of the LMW heparins results in predictable pharmacokinetics and plasma levels in patients with normal renal function. Therefore, LMW heparin levels are not generally measured except in the setting of renal insufficiency, obesity, and pregnancy. LMW heparin levels can be determined by anti-Xa units. For enoxaparin, peak therapeutic levels should be 0.5–1 unit/mL for twice-daily dosing, determined 4 hours after administration, and approximately 1.5 units/mL for once-daily dosing.
Toxicity
A. Bleeding and Miscellaneous Effects
The major adverse effect of heparin is bleeding. This risk can be decreased by scrupulous patient selection, careful control of dosage, and close monitoring. Elderly women and patients with renal failure are more prone to hemorrhage. Heparin is of animal origin and should be used cautiously in patients with allergy. Increased loss of hair and reversible alopecia have been reported. Long-term heparin therapy is associated with osteoporosis and spontaneous fractures. Heparin accelerates the clearing of postprandial lipemia by causing the release of lipoprotein lipase from tissues, and long-term use is associated with mineralocorticoid deficiency.
B. Heparin-Induced Thrombocytopenia
Heparin-induced thrombocytopenia (HIT) is a systemic hypercoagulable state that occurs in 1–4% of individuals treated with UFH for a minimum of 7 days. Surgical patients are at greatest risk. The reported incidence of HIT is lower in pediatric populations outside the critical care setting and is relatively rare in pregnant women. The risk of HIT may be higher in individuals treated with UFH of bovine origin compared with porcine heparin and is lower in those treated exclusively with LMW heparin.
Morbidity and mortality in HIT are related to thrombotic events. Venous thrombosis occurs most commonly, but occlusion of peripheral or central arteries is not infrequent. If an indwelling catheter is present, the risk of thrombosis is increased in that extremity. Skin necrosis has been described, particularly in individuals treated with warfarin in the absence of a direct thrombin inhibitor, presumably due to acute depletion of the vitamin K-dependent anticoagulant protein C occurring in the presence of high levels of procoagulant proteins and an active hypercoagulable state.
The following points should be considered in all patients receiving heparin: Platelet counts should be performed frequently; thrombocytopenia appearing in a time frame consistent with an immune response to heparin should be considered suspicious for HIT; and any new thrombus occurring in a patient receiving heparin therapy should raise suspicion of HIT. Patients who develop HIT are treated by discontinuance of heparin and administration of a direct thrombin inhibitor.
Contraindications
Heparin is contraindicated in patients with HIT, hypersensitivity to the drug, active bleeding, hemophilia, significant thrombocytopenia, purpura, severe hypertension, intracranial hemorrhage, infective endocarditis, active tuberculosis, ulcerative lesions of the gastrointestinal tract, threatened abortion, visceral carcinoma, or advanced hepatic or renal disease. Heparin should be avoided in patients who have recently had surgery of the brain, spinal cord, or eye; and in patients who are undergoing lumbar puncture or regional anesthetic block. Despite the apparent lack of placental transfer, heparin should be used in pregnant women only when clearly indicated.
Administration & Dosage
The indications for the use of heparin are described in the section on clinical pharmacology. A plasma concentration of heparin of 0.2–0.4 unit/mL (by protamine titration) or 0.3–0.7 unit/mL (anti-Xa units) is considered to be the therapeutic range for treatment of venous thromboembolic disease. This concentration generally corresponds to a PTT of 1.5–2.5 times baseline. However, the use of the PTT for heparin monitoring is problematic. There is no standardization scheme for the PTT as there is for the prothrombin time (PT) and its international normalized ratio (INR) in warfarin monitoring. The PTT in seconds for a given heparin concentration varies between different reagent/instrument systems. Thus, if the PTT is used for monitoring, the laboratory should determine the clotting time that corresponds to the therapeutic range by protamine titration or anti-Xa activity, as listed above.
In addition, some patients have a prolonged baseline PTT due to factor deficiency or inhibitors (which could increase bleeding risk) or lupus anticoagulant (which is not associated with bleeding risk but may be associated with thrombosis risk). Using the PTT to assess heparin effect in such patients is very difficult. An alternative is to use anti-Xa activity to assess heparin concentration, a test now widely available on automated coagulation instruments. This approach more accurately measures the heparin concentration; however, it does not provide the global assessment of intrinsic pathway integrity of the PTT.
The following strategy is recommended: prior to initiating anticoagulant therapy of any type, the integrity of the patient’s hemostatic system should be assessed by a careful history of prior bleeding events, and baseline PT and PTT. If there is a prolonged clotting time, the cause of this (deficiency or inhibitor) should be determined prior to initiating therapy, and treatment goals stratified to a risk-benefit assessment. In high-risk patients measuring both the PTT and anti-Xa activity may be useful. When intermittent heparin administration is used, the aPTT or anti-Xa activity should be measured 6 hours after the administered dose to maintain prolongation of the aPTT to 2–2.5 times that of the control value. However, LMW heparin therapy is the preferred option in this case, as no monitoring is required in most patients.
Continuous intravenous administration of heparin is accomplished via an infusion pump. After an initial bolus injection of 80–100 units/kg, a continuous infusion of about 15–22 units/kg/h is required to maintain the anti-Xa activity in the range of 0.3–0.7 units/mL. Low-dose prophylaxis is achieved with subcutaneous administration of heparin, 5000 units every 8–12 hours. Because of the danger of hematoma formation at the injection site, heparin must never be administered intramuscularly.
Prophylactic enoxaparin is given subcutaneously in a dosage of 30 mg twice daily or 40 mg once daily. Full-dose enoxaparin therapy is 1 mg/kg subcutaneously every 12 hours. This corresponds to a therapeutic anti-factor Xa level of 0.5–1 unit/mL. Selected patients may be treated with enoxaparin 1.5 mg/kg once a day, with a target anti-Xa level of 1.5 units/mL. The prophylactic dosage of dalteparin is 5000 units subcutaneously once a day; therapeutic dosing is 200 units/kg once a day for venous disease or 120 units/kg every 12 hours for acute coronary syndrome. LMW heparin should be used with caution in patients with renal insufficiency or body weight greater than 150 kg. Measurement of the anti-Xa level is useful to guide dosing in these individuals.
The synthetic pentasaccharide molecule fondaparinux avidly binds antithrombin with high specific activity, resulting in efficient inactivation of factor Xa. Fondaparinux has a long half-life of 15 hours, allowing for once-daily dosing by subcutaneous administration. Fondaparinux is effective in the prevention and treatment of venous thromboembolism, and does not appear to cross-react with pathologic HIT antibodies in most individuals.
Reversal of Heparin Action
Excessive anticoagulant action of heparin is treated by discontinuance of the drug. If bleeding occurs, administration of a specific antagonist such as protamine sulfate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


