Figure 15–1 Comparison of normal (A) and atherosclerotic (B) arterial walls. Steps in the pathogenesis of atherosclerosis are as follows: (1) Damage to the endothelium is followed by invasion of macrophages. (2) Endothelial and macrophage growth factors stimulate smooth muscle cells to migrate into the tunica intima and to proliferate. (3) Oxidized cholesterol accumulates in and around macrophages (foam cells) and muscle cells. (4) Collagen and elastic fibers form a connective tissue matrix that results in a fibrous plaque.
Because coronary heart disease (CHD) is the main cause of premature death in industrialized countries, it is important to detect and eliminate modifiable risk factors associated with it. In addition to hyperlipidemia, these risk factors include hypertension, cigarette smoking, and a low high-density lipoprotein (HDL) cholesterol level (<40 mg/dL). Unmodifiable risk factors include male gender, family history of premature CHD (CHD in first-degree male relative under 55 years of age or first-degree female under 65 years of age), and advanced age (men over 45 years, women over 55 years). Diabetes mellitus and clinical manifestations of noncoronary forms of atherosclerotic disease (e.g., aortic aneurysm and carotid artery disease) are considered CHD risk equivalents when health care professionals are determining goals for treatment of hyperlipidemia. This means that persons with these conditions are considered to have the same risk for future occurrences of CHD as persons who already have some form of CHD (e.g., angina pectoris, myocardial infarction, or other types of clinically significant myocardial ischemia).
Women have a lower risk of heart disease until after menopause, and this lower risk may be partly owing to the favorable effect of estrogens on serum lipoprotein levels. Estrogens may also have beneficial effects on the microcirculation and energy metabolism.
After discussing lipoproteins, lipid transport, and the causes and types of hyperlipoproteinemia, this chapter describes how dietary restrictions, alone or in combination with drug treatment, can reduce serum lipoprotein levels and thereby reduce the risk of CHD.
Lipoproteins and Lipid Transport
Because lipids are insoluble in plasma, they must be transported in the circulation in the form of lipoproteins. There are numerous types of lipoproteins, including chylomicrons, very low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs), intermediate-density lipoproteins, HDLs, and lipoprotein(a). The various types are distinguished in terms of their buoyant density, lipid and protein composition, and role in lipid transport. Moreover, each type is associated with a unique group of apoproteins. Some of the apoproteins are exchanged between different types of lipoproteins as they transport lipids to various tissues. The composition and metabolism of lipoproteins is depicted in Box 15–1.
BOX 15–1 LIPOPROTEIN METABOLISM AND ATHEROSCLEROSIS
The liver is the central processing site for lipoprotein metabolism. Cholesterol is derived from three sources: (1) biosynthesis from acetyl-CoA, (2) delivery of dietary cholesterol by chylomicron remnants, and (3) endocytosis of LDL cholesterol by LDL receptors.
Triglycerides are formed in the liver from fatty acids, which are derived from lipolysis of triglycerides in adipose tissue. Triglycerides, cholesterol, and protein are packaged by Golgi bodies in the liver to form VLDLs, which are secreted into the circulation. The VLDLs accept apoproteins C and E from HDLs and then return these apoproteins to HDL as they deliver triglycerides (as fatty acids) to adipose and other tissues via the action of lipoprotein lipase located in the capillary endothelium.
| Lipoprotein | Core Lipids∗ | Apoproteins∗ |
|---|---|---|
| Chylomicron | Dietary triglycerides and cholesteryl esters | B-48, C, E, and A |
| VLDL | Endogenous triglycerides and cholesteryl esters | C, B-100, and E |
| LDL | Cholesteryl esters | B-100 |
| HDL | Cholesteryl esters and phospholipids | A-I, A-II, C, E, and D |
∗ Listed in order of quantitative importance.
With the removal of apoproteins and triglycerides, VLDLs are transformed into LDLs and intermediate-density lipoproteins. The LDLs deliver cholesterol to various sites in the body, including peripheral tissues and the liver via endocytosis by LDL receptors. LDL cholesterol is also incorporated into atherosclerotic plaques.
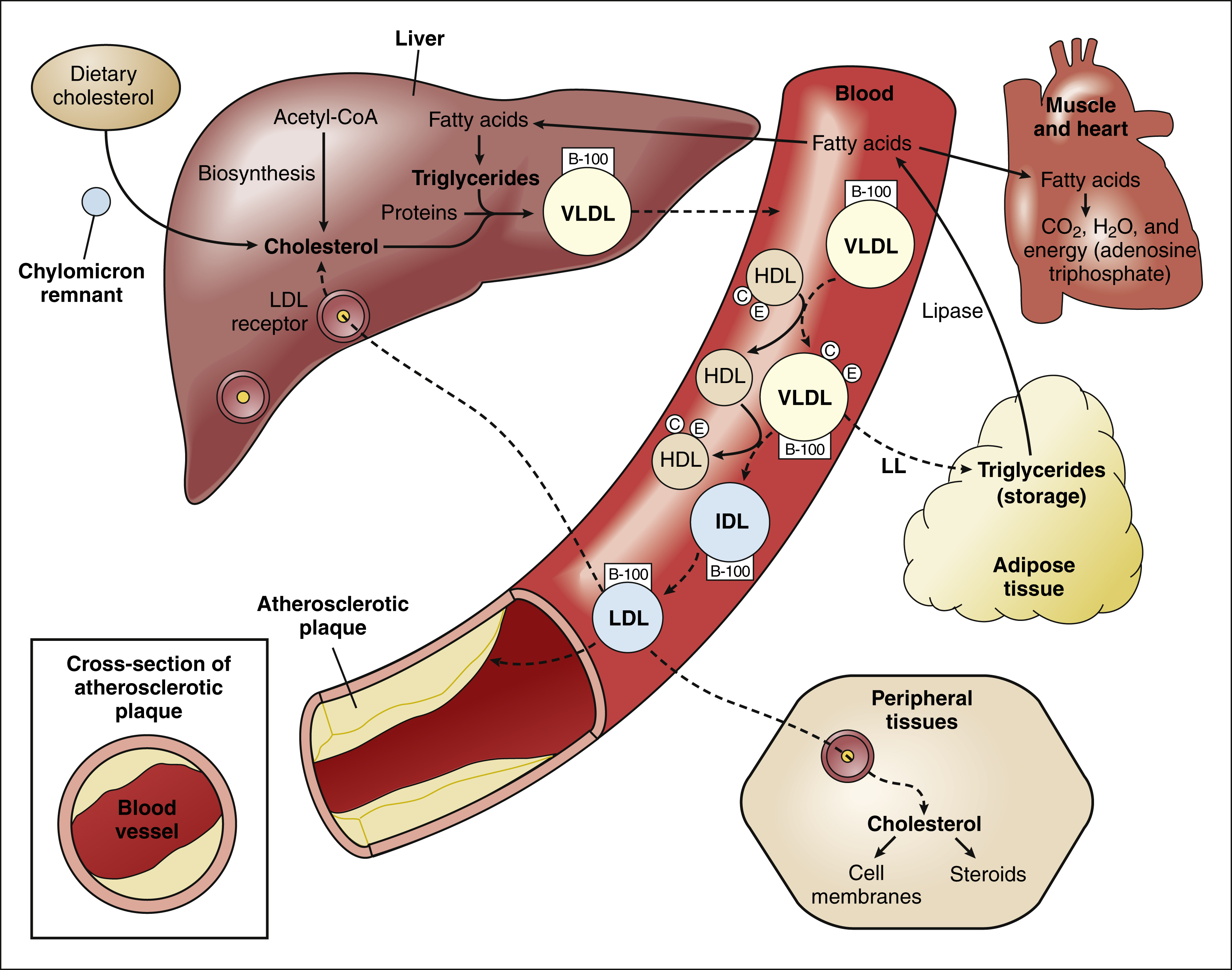
Chylomicrons
Chylomicrons are involved primarily in the transport of dietary lipids from the gut to the adipose tissue and liver. When cholesterol and triglycerides are ingested, they are emulsified in the intestines by the bile acids and other bile secretions, and the emulsified lipids are combined with proteins to form chylomicrons in the gut wall. After chylomicrons are secreted into the circulation, they deliver triglycerides to adipose tissue via the action of a lipoprotein lipase located in the vascular endothelial cells. By this process, chylomicrons are converted to a cholesterol-rich chylomicron remnant, which transports cholesterol to the liver.
Very Low-Density and Low-Density Lipoproteins
Golgi bodies in the liver form VLDLs from triglycerides, cholesterol, and protein and then secrete the VLDLs into the circulation. The VLDLs deliver triglycerides to adipose tissue in the same manner as do the chylomicrons. During the process, the VLDLs are transformed into intermediate-density lipoproteins and LDLs that contain a high percentage of cholesterol.
The LDLs transport cholesterol to peripheral tissues for incorporation into cell membranes and steroids. In this process, the LDLs bind to specific LDL receptors that are located in the plasma membrane of cells and recognize apoprotein B-100 on the surface of LDL molecules. After binding to their receptors, the LDLs undergo endocytosis and are incorporated into lysosomes for further processing of cholesterol and protein.
The LDLs can also deliver cholesterol to nascent atheromas and thereby contribute to the development of atherosclerosis (see Fig. 15–1). In atheromas, cholesterol is phagocytosed by macrophages, which are transformed into foam cells as they become filled with oxidized cholesterol.
High-Density Lipoproteins
The HDLs are small lipoproteins whose high density is caused by their high ratio of protein to lipid. Nascent pre-β-HDL particles are formed in the liver and intestines from apoprotein A-I and a small quantity of cholesterol and phospholipid. The cholesterol is then esterified by lecithin-cholesterol acyltransferase so as to convert pre-β-HDL into mature α-HDL particles. As HDL circulates in the blood, it exchanges apoproteins C and E with VLDL, so as to enable delivery of VLDL triglycerides to adipose tissue via lipoprotein lipase.
HDL transports cholesterol from atheromas and peripheral tissues to the liver. During this process of reverse cholesterol transport, the α-HDLs acquire additional cholesterol from macrophages in blood vessel walls (via adenosine triphosphate–binding cassette transporters) and esterify the cholesterol via lecithin-cholesterol acyltransferase. The cholesteryl esters are either transported by HDL directly to the liver or are transferred to LDL for transport to the liver (indirect pathway). The contribution of reverse cholesterol transport to CHD has been supported by epidemiologic studies that show an inverse correlation between HDL levels and the risk of this disease.
Lipoprotein(a)
Lipoprotein(a) is a unique lipoprotein whose physiologic function is unknown and whose occurrence is genetically determined. It is found in atherosclerotic plaques of some individuals, and plasma levels of lipoprotein(a) are highly correlated with angiographically demonstrable coronary artery disease.
Causes and Types of Hyperlipoproteinemia
Hyperlipoproteinemia occurs as a result of genetic or environmental factors that increase the formation of lipoproteins or reduce the clearance of lipoproteins from the circulation. These factors include biochemical defects in lipoprotein metabolism, excessive dietary intake of lipids, endocrine abnormalities, and use of drugs that perturb lipoprotein formation or catabolism. Table 15–1 provides information about the characteristics and types of hyperlipoproteinemia.
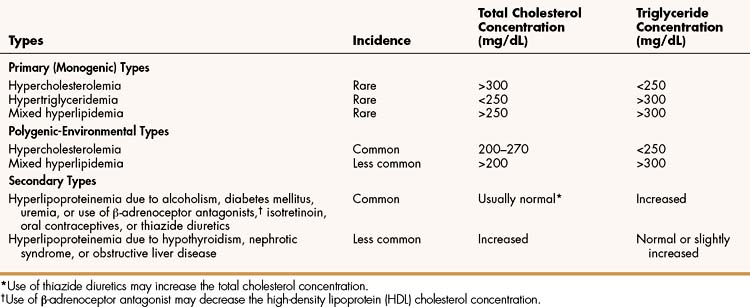
Primary hyperlipoproteinemias are relatively rare disorders, each of which is caused by a monogenic defect (a specific defect at a single gene). In some disorders, LDL cholesterol (LDL-C) levels are severely elevated because of a deficiency of LDL receptors or a defect in the structure of apoprotein B. In the latter case, LDL receptors do not recognize LDL, so LDL removal from the circulation is markedly impaired. In another disorder, VLDL and triglyceride levels are severely elevated because of a lipoprotein lipase deficiency that prevents delivery of triglycerides to adipose tissue.
Most cases of hyperlipoproteinemia do not result from a single gene defect but instead result from the influence of several genes that predispose the patient to milder forms of hyperlipoproteinemia, particularly in the presence of excessive dietary intake of lipids. These milder forms, called polygenic-environmental hyperlipoproteinemias, which are much more common than primary hyperlipoproteinemias, are responsible for most cases of accelerated atherosclerosis.
Secondary hyperlipoproteinemias are commonly caused by the presence of alcoholism, diabetes mellitus, or uremia or by the use of drugs such as β-adrenoceptor antagonists, isotretinoin, oral contraceptives, or thiazide diuretics. They are less commonly caused by hypothyroidism, nephrotic syndrome, or obstructive liver disease.
Guidelines for Management of Hypercholesterolemia
The Adult Treatment Panel III of the National Cholesterol Education Program (NCEP) issued evidence-based guidelines for the management of high blood cholesterol and related disorders in 2001. These recommendations were updated in 2004 based on evidence from five clinical trials published since the Adult Treatment Panel III guidelines were issued. These trials confirmed the benefits of cholesterol-lowering therapy in high-risk patients and encourage more aggressive reductions in LDL-C in very high-risk patients.
The NCEP guidelines (Table 15–2) establish LDL-C goals and levels for initiating therapeutic lifestyle changes (TLCs) and drug therapy for persons in different risk categories. For high-risk patients (who already have CHD or have CHD risk equivalents), the basic goal is to achieve a LDL-C of less than 100 mg/dL, and TLC and drug therapy should be initiated if the patient’s LDL-C is higher than 100 mg/dL. The updated guidelines suggest an optional LDL-C goal for high-risk patients of less than 70 mg/dL, particularly for those whose LDL-C is less than 100 mg/dL at baseline. This optional goal is based on clinical trials that show that high-risk patients benefit from LDL-C reduction regardless of their baseline level.
TABLE 15–2 National Cholesterol Education Program Guidelines for Management of High Blood Cholesterol Levels for Persons in Different Risk Categories∗
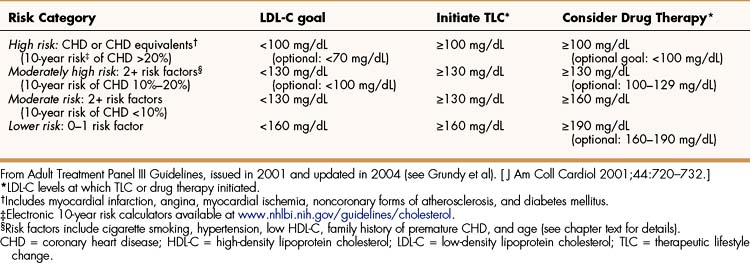
For moderately high-risk patients (persons with two or more CHD risk factors and a 10% to 20% risk of developing CHD in 10 years), the original guidelines suggest a LDL-C goal of less than 130 mg/dL, with TLC and drug therapy initiated if levels are higher than 130 mg/dL. The updated guidelines recommend considering drug therapy for patients with LDL-C levels between 100 and 129 mg/dL. The 10-year risk of developing CHD can be estimated using calculators such as the one provided by the National Heart, Lung, and Blood Institute (see Table 15–2).
Patients with a moderate risk of CHD (two or more risk factors and <10% risk of developing CHD in 10 years) have the same LDL-C goal and TLC initiation level as moderately high-risk patients, but drug therapy is recommended only at the start of therapy if LDL-C levels are above 160 mg/dL. If TLCs do not achieve the target level in 6 to 12 months, drug therapy should be considered for persons with a moderate risk of CHD.
For persons with a lower risk of CHD (zero to one risk factor), the target LDL-C level is less than 160 mg/dL. For this reason, TLC should be initiated in persons exceeding this level. The original guidelines recommended drug therapy only at the start of treatment if LDL-C levels exceed 190 mg/dL, but the updated recommendations suggest considering drug therapy if levels are between 160 and 189 mg/dL.
The updated guidelines also suggest that high-risk patients with high triglycerides or a low HDL-cholesterol (HDL-C) level receive niacin or a fibrate drug in combination with a LDL-C lowering drug to raise HDL-C levels and lower triglyceride levels. This is because epidemiologic studies show a correlation between CHD and high levels of plasma triglycerides or low levels of HDL.
Secondary causes of hyperlipidemia should be excluded before treatment is considered, because abnormal levels of cholesterol or triglycerides can often be corrected by proper management of the underlying condition or by replacing the offending drug with an alternative.
Therapeutic Lifestyle Changes
TLCs are an essential modality in the management of high cholesterol and triglyceride levels and may be effective by themselves in patients with mildly elevated cholesterol or triglyceride levels. The TLC modality includes recommendations for diet, weight management, and physical activity. The diet of patients with hypercholesterolemia should be low in cholesterol, saturated fat, and calories. Saturated fat and cholesterol are restricted because each independently increases LDL-C, and calories are restricted to help the patient achieve or maintain an ideal body weight. The Adult Treatment Panel III guidelines recommend the American Heart Association step II diet as the initial diet for lowering cholesterol and triglyceride levels and suggest adding plant stanol or sterol esters, also called phytosterols (2 g/day), and soluble fiber (10 to 25 g/day) to enhance LDL-C reduction.
The American Health Association step II diet mandates that cholesterol intake should be under 200 mg/day, and total calories from fat should be limited to 25% to 35% of total calories, with saturated fat limited to less than 7% of total calories. Foods containing partially hydrogenated plant oils should be avoided because hydrogenation produces the trans isomers of fatty acids that acts to increase LDL-C levels, decrease HDL-C levels, increase systemic inflammation, and reduce the availability of fatty acid precursors to anticoagulant prostaglandins.
Dietary modulations are particularly useful in persons with multiple risk factors or angiographic evidence of coronary artery disease. Studies have shown that diets low in saturated and trans fatty acids and high in linoleic acid and omega-3 fatty acids (linolenic acid and those in fish oils) improve the LDL-C to HDL-C ratio. These diets may also reverse the angiographic evidence of coronary atherosclerosis and reduce mortality rate in patients with CHD. In patients with hypertriglyceridemia, supplementing the diet with fish oils that contain omega-3 fatty acids often lowers triglyceride levels. The omega-3 fatty acids contain a double bond between the third and fourth carbon from the end of the molecule.
DRUGS FOR HYPERCHOLESTEROLEMIA
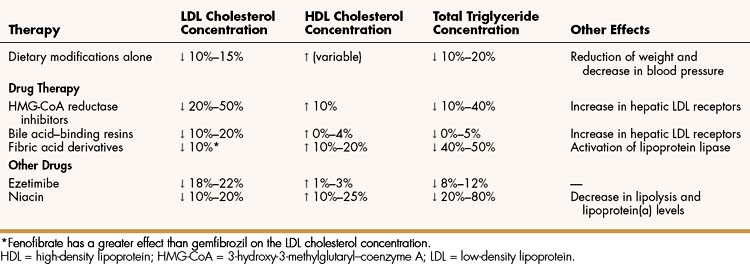
The 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase inhibitors (statins), the bile acid–binding resins, and ezetimibe are used primarily to treat hypercholesterolemia, whereas the fibric acid derivatives and niacin are used to reduce elevated triglyceride levels and to raise HDL levels. Table 15–3, Table 15–4, and Table 15–5 outline the effects and pharmacokinetic properties of the drugs used to treat hypercholesterolemia and compare them with the effects and properties of other drugs discussed in this chapter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


