Baran A. Ersoy and Keith A. Hoffmaster
Concentrative and Equilibrative Nucleoside Transporter (CNT and ENT) Families |
INTEGRATION OF DRUG TRANSPORTER SCIENCE INTO DRUG DISCOVERY AND DEVELOPMENT |

Membrane transporters are proteins that facilitate the transport of soluble small molecules across lipid bilayers. These transporters regulate the tissue and plasma distribution as well as the excretion of drugs and endogenous compounds and therefore affect their pharmacokinetic profile (i.e., absorption, distribution, metabolism, and excretion). Transporters are generally classified as uptake or efflux proteins depending on the direction in which they move substrates across the membranes of cells. They include several protein families that exhibit broad substrate specificity and tissue distribution. The function of transporters depends on the tissue in which they are expressed as well as their subcellular localization to the apical or basolateral membrane (Fig. 5-1). Whereas transporters expressed in organs of elimination (e.g., liver or kidney) can facilitate the clearance of drugs and/or metabolites from the systemic circulation, efflux transporters in non-eliminating organs such as the central nervous system and placenta primarily function to minimize the exposure of the brain and the developing fetus, respectively, to potentially harmful exogenous substrates. In some cases, intestinal efflux transporters expressed at the apical membrane of the enterocyte can limit the oral bioavailability of drugs; in other cases, uptake transporters expressed at this membrane can aid in the systemic absorption of compounds, thereby enhancing their oral bioavailability. Transporter function in the liver often facilitates drug clearance, but efficient active uptake into the liver after an oral dose can also affect drug bioavailability by contributing to first-pass hepatic metabolism and elimination. Because drugs can both inhibit and induce transporters, concurrent administration of multiple drugs may alter the pharmacokinetics and pharmacodynamics of the other compounds depending on transporter affinity, specificity, and alternate mechanisms of drug disposition. Therefore, identifying the specific transporter(s) for which a novel drug is a substrate (or inhibitor) may not only help to devise optimization and formulation strategies that improve drug bioavailability and efficacy, but may also prevent adverse effects by anticipating and limiting drug–drug interactions. There are over 400 transporters in the human genome. This chapter provides an overview of this effective but complex system by focusing on a select number of well-characterized transporters involved in drug disposition and in the flux of endogenous substrates and on those that may become the sites of drug–drug interactions.
 Mr. H is a 47-year-old mildly overweight man who comes to the walk-in clinic complaining of muscle pain and weakness in his arms and legs. He has been taking a statin medication for the past 4 years and has been able to manage his cholesterol levels effectively with this medication and diet modifications. When asked about any recent changes to his medications or diet, he indicates that his primary care physician started him 3 weeks ago on medical therapy to help control his triglyceride levels. In addition to the recent muscle pain, Mr. H has noticed a mild rash on his torso over the past 3 weeks since starting the new medication prescribed to help control his triglycerides.
Mr. H is a 47-year-old mildly overweight man who comes to the walk-in clinic complaining of muscle pain and weakness in his arms and legs. He has been taking a statin medication for the past 4 years and has been able to manage his cholesterol levels effectively with this medication and diet modifications. When asked about any recent changes to his medications or diet, he indicates that his primary care physician started him 3 weeks ago on medical therapy to help control his triglyceride levels. In addition to the recent muscle pain, Mr. H has noticed a mild rash on his torso over the past 3 weeks since starting the new medication prescribed to help control his triglycerides.
Questions
1. Without knowing the specific medications that Mr. H is taking, which drug–drug interactions do you suspect might be contributing to his new symptoms?
2. Consultation with Mr. H’s primary care physician reveals that Mr. H is also taking a fixed-dose combination of lopinavir/ritonavir to manage his HIV infection. How might this complicate the drug–drug interaction suspected in question 1?
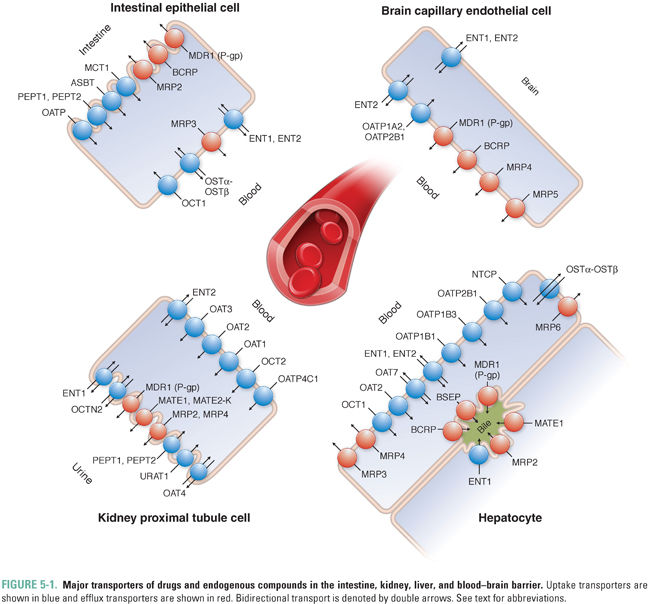
The Human Genome Organisation (HUGO) Gene Nomenclature Committee (HGNC) has approved standard acronyms for both solute carrier (SLC) and ATP-binding cassette (ABC) families of transporters. However, many drug transporters, especially those discussed in greater detail in this chapter, were initially cloned and named based on their pharmacologic attributes such as substrate specificity and association with drug resistance. For instance, the ABCB1 gene was originally named and is more commonly referred to as P-glycoprotein (P-gp) because it was initially identified as a glycoprotein that controls cell membrane permeability (P). Ensuing studies have also led to its characterization as a multidrug resistance (MDR) protein encoded by the MDR1 gene. Therefore, P-gp, MDR1, and ABCB1 are all synonyms for the same transporter. In this chapter, the transporters are referred to using popular names, with HGNC nomenclature and other acronyms stated in parentheses. Additional details on nomenclature and synonyms/previous names can be found at http://www.genenames.org/genefamilies/ABC and http://www.genenames.org/genefamilies/SLC.
 UPTAKE AND EFFLUX TRANSPORTERS
UPTAKE AND EFFLUX TRANSPORTERS
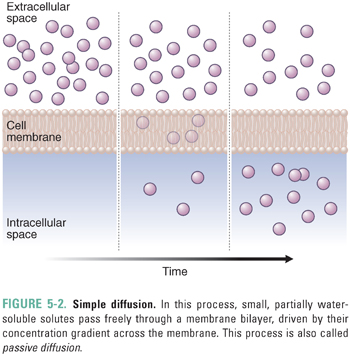
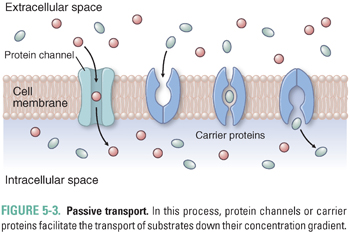
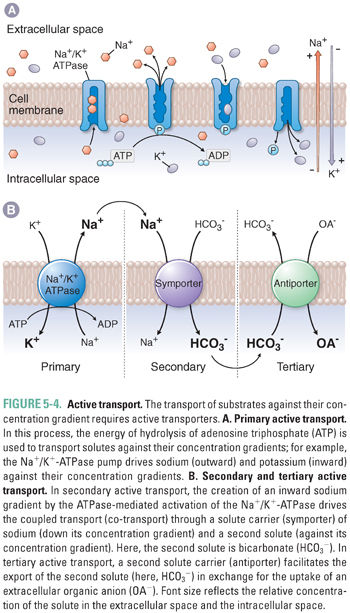
Drugs and endogenous compounds cross cellular membrane barriers by simple diffusion, passive transport, or active transport. Simple diffusion (also called passive diffusion) can occur when small, partially water-soluble solutes such as certain polar lipids pass freely through a membrane bilayer, driven by their concentration gradient across the membrane (Fig. 5-2). However, many molecules require the assistance of membrane transporters to move across membranes. Solute carriers facilitate the passive transport of substrates down their concentration gradient (Fig. 5-3). In contrast, the transport of substrates against their concentration gradient requires active transporters (Fig. 5-4). Primary active transport utilizes the energy of hydrolysis of adenosine triphosphate (ATP); the substrate first binds to the transporter, and substrate transport across the membrane is completed upon ATP-mediated activation of the transporter (Fig. 5-4A). Secondary active transport depends on the creation of an inward sodium gradient by the ATP-mediated activation of the Na+/K+-ATPase pump. The inward sodium gradient drives the coupled transport (co-transport) through a solute carrier of sodium (down its electrochemical gradient) and of a second solute (against its concentration gradient) (Fig. 5-4B). This mechanism is called secondary active transport because the transport through the solute carrier does not directly require ATP hydrolysis but is coupled to a primary ATPase. Tertiary active transport also requires an ATPase-dependent sodium gradient. However, in this case, sodium influx facilitates the diffusion and intracellular accumulation of an anion such as bicarbonate via a secondary active transporter. In the final step of tertiary active transport, the solute carrier exports the intracellular anion in exchange for the uptake of an extracellular organic anion (Fig. 5-4B). With the exception of OSTα-OSTβ (which transports bile acids; see below), all known uptake transporters lack ATPase activity and belong to the solute carrier (SLC) family. Secondary or tertiary active transport mechanisms are required for cellular absorption of many drugs, nutrients, and endogenous metabolites. The next section outlines the transporters that have essential roles in the uptake of drugs and endogenous compounds in key tissues that are involved in the absorption, distribution, and elimination of drugs. These tissues include the intestine, liver, kidney, and endothelial cells of the blood–brain barrier.
Organic Anion-Transporting Polypeptide (OATP) Family
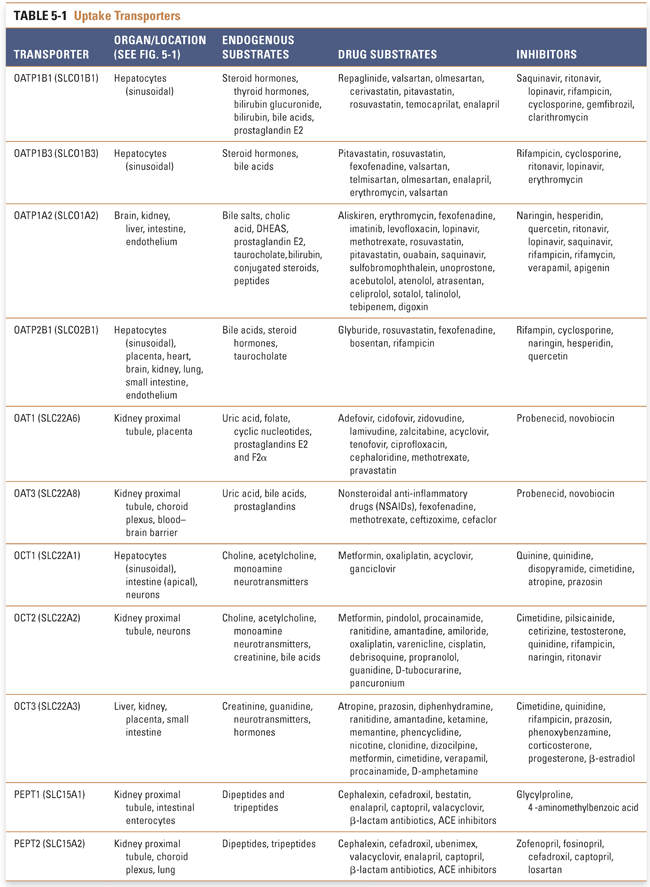
OATPs (SLCO, former SLC21) are expressed in all epithelial cells. These transporters facilitate the uptake of large hydrophobic and amphiphilic organic compounds, such as bile acids, thyroid hormones, conjugated steroids, and eicosanoids (Table 5-1). Although OATPs are primarily responsible for the transport of anionic compounds, some members of the OATP family transport bulky type II organic cations such as rocuronium. The accumulation of substrates such as bile acids within the liver is a concentrating process, and OATPs move substrates into cells by a tertiary active transport mechanism. Of the 11 family members, 5 have been implicated in the transport of xenobiotics. OATP1B1, OATP1B3, and OATP2B1 mediate the uptake of drugs (e.g., statins) across the sinusoidal membrane of hepatocytes, whereupon the drugs can be metabolized by enzymes such as cytochrome P450s (CYPs) and/or secreted into the bile or back into the systemic circulation (Fig. 5-1). Inhibition of hepatic OATPs has been implicated as a potential mechanism of drug–drug interactions. The potential for these interactions has resulted in revised dosing guidelines for some statins when they are administered with drugs that inhibit OATPs (e.g., cyclosporine, gemfibrozil, lopinavir/ritonavir). OATP1A2 is ubiquitously expressed and contributes primarily to drug absorption from the intestinal lumen into intestinal epithelial cells. OATP4C1 facilitates the uptake of drugs such as digoxin from the circulation into kidney proximal tubule cells, from which the drugs are eliminated via the urine (Fig. 5-1).
In the introductory case, Mr. H was most likely started on a drug that inhibited the hepatic uptake of his statin, and thereby increased the systemic bioavailability of the statin, via a transporter-related drug–drug interaction. Fibrates such as gemfibrozil are commonly used to reduce triglyceride levels when statins and diet modification prove insufficient. Concurrent administration of statins with gemfibrozil can cause myopathy, however, in part due to inhibition of OATP1B1-mediated hepatic uptake of statins by gemfibrozil. This drug–drug interaction results in increased blood levels of the statin and consequent systemic toxicity. Mr. H was also taking a fixed dose of lopinavir/ritonavir, which is an inhibitor of OATP1B1- and OATP1B3-mediated drug uptake into the liver; this combination of HIV protease inhibitors may have further contributed to drug–drug interactions and exacerbated systemic adverse effects of Mr. H’s statin after initiation of treatment for hypertriglyceridemia.
Organic Anion Transporter (OAT) Family
OATs, which belong to the SLC22A family, mediate the cellular uptake of small organic anions such as conjugated steroids, biogenic amines, and cGMP as well as a broad range of xenobiotics such as antivirals, antibiotics, ACE inhibitors, and anticancer drugs (Table 5-1). Despite their classification as uptake transporters, OAT isoforms 1–4 and 7 have essential roles in drug clearance by facilitating the uptake of drugs from the systemic circulation into the liver and kidneys, where they can be metabolized and excreted (Fig. 5-1). Uptake of anions into cells against their electrochemical gradient requires OATs to function as tertiary active transporters. OAT1, OAT3, and OAT4 exchange intracellular 2-oxoglutarate, and OAT7 exports intracellular short-chain fatty acids such as butyrate in exchange for their extracellular substrates. OAT1, OAT2, and OAT3 clear many organic anions from the systemic circulation into the kidney proximal tubule, from which the anions are eliminated in the urine. OAT2 and OAT7 are expressed mainly on the sinusoidal membrane of hepatocytes. Unlike OAT1, OAT2, and OAT3, OAT4 and the urate anion exchanger 1 (URAT1) are expressed on the apical (brush border) membrane of the kidney proximal tubule, where they mediate the reabsorption of uric acid from urine. Therefore, drugs that inhibit OAT4 and URAT1 may decrease blood uric acid levels and thereby provide therapeutic benefit (e.g., in the treatment of gout by the URAT1 inhibitor probenecid; see Chapter 49, Integrative Inflammation Pharmacology: Gout) and may potentially cause enhanced elimination of OAT4 substrates.
Organic Cation Transporter (OCT) Family
Like OATs, OCTs belong to the SLC22A family and contribute to the renal clearance of xenobiotics such as antiviral drugs. OCTs also mediate the transport of a diverse group of small organic cations such as catecholamines, hormones, and neurotransmitters (Table 5-1). Transport of cations by OCTs occurs down the electrochemical gradient of the solute and does not depend on ATP hydrolysis or ion exchange; instead, the transport is thought to be driven by differences in membrane potential. OCT isoforms can have overlapping substrates, and the transport of solutes can be bidirectional depending on the electrochemical gradient. There is strong evidence for the roles of OCT1, OCT2, and OCT3 in drug disposition (Table 5-1; Fig. 5-1). OCT1 is highly expressed in the sinusoidal (basolateral) membrane of hepatocytes. OCT2 is expressed mostly in the kidney proximal tubule and it contributes to the uptake of metabolites from the blood into the tubule. In contrast, OCT3 exhibits broad tissue distribution; its highest expression is in the intestine, liver, and kidney, where it facilitates intestinal absorption and hepatic and renal secretion of drugs, respectively. All three transporters mediate the uptake of a wide array of therapeutic agents, including sedatives, antidepressants, β-blockers, and antidiabetic drugs such as metformin. OCTs are also an important site of drug–drug interactions. In certain cases, OCT-mediated renal uptake can contribute to the adverse effects of nephrotoxic drugs, which can be prevented by the concomitant administration of an OCT inhibitor.
A significant fraction of bile acids are recycled via three main transporter mechanisms in the liver and gastrointestinal (GI) tract. Na+/taurocholate co-transporting polypeptide (NTCP, SLC10A1) is exclusively expressed at the sinusoidal membrane of hepatocytes and is a key mechanism in the transport of conjugated and unconjugated bile acids from the circulation into the liver (Fig. 5-1). Whereas OATPs are responsible for the sodium-independent uptake of bile acids, NTCP is responsible for sodium-dependent secondary active bile acid transport that is coupled to the activation of Na+/K+-ATPase. In addition to bile acids, NTCP mediates partial uptake of some statins (e.g., rosuvastatin; Table 5-1). The apical sodium-dependent bile acid transporter (ASBT, SLC10A2) is expressed at the apical membrane of the epithelial cells of the distal small intestine and mediates the uptake of bile acids from the intestinal lumen (Fig. 5-1, Table 5-1). OSTα-OSTβ is the only uptake transporter that does not belong to the SLC family. It comprises a heterodimer of two different subunits and can function as either an efflux transporter or an uptake transporter depending on the electrochemical gradient of its substrates. However, the major function of the transporter is to contribute to the enterohepatic recirculation of bile acids: OSTα-OSTβ mediates the efflux of bile acids and conjugated steroids from intestinal epithelia into the circulation, from which the bile acids are taken up by hepatocytes (Fig. 5-1, Table 5-1).
Peptide Transporter (PEPT) Family
PEPT family transporters (SLC15A) are proton-driven symporters that are highly expressed in the intestine and kidney. PEPT1 has a key role in the absorption of dietary nitrogen, in the form of dipeptides and tripeptides, from the lumen of the small intestine into enterocytes. Peptide-like metabolites or drugs such as β-lactam antibiotics and ACE inhibitors exhibit high bioavailability due largely to PEPT1-mediated absorption (Table 5-1). Both PEPT1 and PEPT2 are expressed in the kidney, where they mediate reuptake of small peptides at the apical membrane of the kidney proximal tubule and regulate systemic nitrogen balance (Fig. 5-1).
Concentrative and Equilibrative Nucleoside Transporter (CNT and ENT) Families
CNT (SLC28) family members CNT1, CNT2, and CNT3 mediate sodium-dependent uptake of nucleosides by epithelial cells. CNT1 and CNT2 specifically transport pyrimidine and purine nucleosides, respectively, whereas CNT3 is capable of transporting both classes of nucleosides. In contrast, ENT (SLC29) family members provide bidirectional transport of purine and pyrimidine nucleosides. The direction of ENT-mediated transport depends on the concentration gradient of the nucleosides and functions to equilibrate extracellular and intracellular nucleoside levels. Whereas intracellular nucleoside accumulation contributes to nucleotide synthesis, CNT- and ENT-mediated nucleoside uptake may also limit the activation of extracellular nucleoside signaling events, such as the activation of adenosine receptors. In addition to endogenous nucleosides, CNTs and ENTs transport nucleoside analogues such as cytotoxic anticancer (e.g., gemcitabine) and antiviral (e.g., zidovudine) drugs. Therefore, factors or compounds that reduce the expression or activity of these transporters on target tissues may reduce the efficacy of nucleoside anticancer and antiviral drugs.
The glucose transporter family (GLUT, SLC2) regulates the distribution of glucose between the plasma and tissues. Among the several family members, GLUT1–4 isoforms are the most studied and most relevant to glucose metabolism. GLUT1 has broad tissue expression and is responsible for sustaining basal cellular glucose levels with a slow uptake rate. GLUT2 is expressed in the organs that participate in the regulation of plasma glucose levels, such as kidney, liver, intestine, and pancreas. GLUT2 mediates the uptake of dietary glucose in the intestine. Because it has relatively low affinity for glucose, GLUT2 acts as a sensor for glucose levels within the pancreatic beta cells that secrete insulin in response to increases in plasma glucose. GLUT2 exhibits bidirectional transport capability, allowing for the influx or efflux of glucose across the cell membrane. The efflux transporter activity of GLUT2 at the sinusoidal membrane of hepatocytes is essential for the transport of glucose produced in the liver into the plasma in order to maintain plasma glucose homeostasis during starvation or fasting. GLUT3 is enriched in neurons; it exhibits high relative affinity for glucose in order to provide a constant influx of glucose from the circulation into neuronal cells, even at low plasma glucose concentrations. GLUT4 mediates insulin-sensitive glucose uptake and storage in the adipose tissue and striated muscle. Under conditions of low plasma insulin concentrations, such as fasting, GLUT4 is sequestered in intracellular vesicles and does not transport glucose. Insulin stimulation causes GLUT4 to translocate to the plasma membrane, where it initiates glucose uptake (see Fig. 31-4).
Sodium glucose transport protein (SGLT, SLC5) family members 1 and 2 are symporters that transport glucose against its concentration gradient via a secondary active mechanism. SGLT1 at the intestinal brush border and SGLT1 and SGLT2 at the kidney proximal tubule brush border mediate glucose absorption from the intestinal lumen and the renal filtrate, respectively. In turn, GLUT2 efflux activity at the apical membrane of these tissues facilitates glucose transport from the cells into the circulation. SGLT2 is responsible for more than 90% of glucose reuptake from the renal filtrate, and inhibition of SGLT2 reduces plasma glucose levels. SGLT2 has been targeted for pharmacologic intervention in the management of diabetes. Dapagliflozin selectively inhibits SGLT2 over SGLT1; this selective inhibition facilitates reduction of plasma glucose levels without impairing SGLT1-mediated intestinal glucose absorption, potentially limiting adverse effects such as diarrhea that may be associated with elevated intestinal glucose levels.
Oral drug administration generally provides the most convenient and affordable route for the systemic delivery of therapeutic agents; high, reproducible oral bioavailability minimizes variability in drug exposure across patient populations. After oral administration, drugs typically are absorbed from the intestinal lumen, delivered into the mesenteric blood supply, and then circulated through the hepatic portal system via the portal vein before entering the systemic circulation (see Chapter 3, Pharmacokinetics). Efflux transporters can affect oral bioavailability (1) by limiting the amount of drug that is absorbed across the enterocyte and/or (2) by transporting drug from the liver into the bile and thereby contributing to a first-pass effect. Whereas efflux transporters expressed on the canalicular membrane of the hepatocyte facilitate the excretion of xenobiotics into the bile, efflux transporters expressed on the sinusoidal (basolateral) membrane can promote the flux of drug and metabolites back into the systemic circulation (Fig. 5-1). Compounds eliminated into the bile across the canalicular membrane of the hepatocyte are concentrated in the gall bladder and released into the small intestine, where both parent drug and possibly metabolites can be absorbed across the intestine (a process known as enterohepatic recirculation) or eliminated into the feces. However, the same first-pass mechanisms (i.e., absorption, efflux, metabolism, and biliary excretion) that limited the compound’s ability to reach the systemic circulation and the peripheral tissues still exist. The oral bioavailability of compounds that undergo extensive enterohepatic recirculation may appear extremely low; however, in cases where the target tissue is either the liver (e.g., statins) or intestine, these compounds may have desired pharmacologic effects despite limited systemic exposure. For drugs that are high-affinity substrates for intestinal and hepatic transporters, alternative routes of administration (such as intravenous and subcutaneous) can circumvent the first-pass effect and possibly increase the systemic exposure. However, even when drug delivery is used to avoid a first-pass effect, the drugs that reach the systemic circulation must nevertheless transit through the hepatobiliary system and may still be cleared rapidly due to efficient drug efflux processes. Compounds in the systemic circulation can also be eliminated in the kidney via active efflux into the urine. Because the transport of compounds from within the intracellular space is often against a concentration gradient, the process requires active transport; the majority of efflux pumps belong to the ATP-binding cassette (ABC) family of active transporters. This section outlines the efflux transporters that have major roles in drug disposition as well as those that facilitate the transport of endogenous compounds.
ATP-Binding Cassette (ABC) Transporter Family
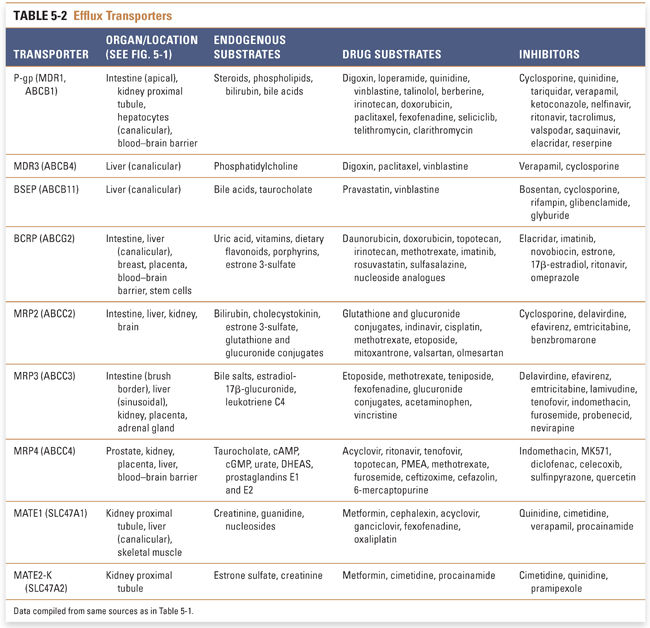
ABC transporters constitute the largest transporter superfamily. They are active transporters that mediate efflux of various substrates—such as phospholipids, steroids, and drugs—out of cells against their concentration gradient. The superfamily is divided into seven families, of which only members of the ABCB, ABCC, and ABCG families have essential roles in drug disposition. ABCA and ABCD proteins transport only endogenous substrates and regulate cellular cholesterol and fatty acid metabolism, and ABCE and ABCF family members lack transmembrane domains and are not involved in the transport of drugs or endogenous compounds across the membranes of cells.
P-Glycoprotein and Bile Salt Export Pump (ABCB Family)
The ABCB family, also known as multidrug resistance/transporters associated with antigen processing (MDR/TAP), consists of 11 members and includes perhaps the most studied drug transporter, P-glycoprotein (P-gp, also known as MDR1 or ABCB1). P-gp was initially discovered and characterized as a protein that mediates anticancer drug resistance. Its expression is elevated in many cancer cells, and P-gp contributes to multidrug resistance against antineoplastic therapeutics. P-gp is also expressed in the apical membrane of the small intestine, liver, kidney, endothelial cells of the blood–brain barrier, and placenta (Fig. 5-1), and it both limits exposure of substrates to certain organs and serves as a mechanism to eliminate xenobiotics from the body. P-gp has broad substrate specificity and it exhibits high affinity for cationic and amphiphilic compounds such as phospholipids (Table 5-2).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


