Michael W. Conner, Catherine Dorian-Conner, Vishal S. Vaidya, Laura C. Green, and David E. Golan

Like many medical interventions, the use of drugs for therapeutic benefit is subject to the law of unintended consequences. These consequences—termed side effects, adverse effects, or toxic effects—are a function of the mechanisms of drug action, the size of the drug dose, and the characteristics and health status of the patient. As such, the principles of pharmacology, presented in the preceding chapters, apply to drug toxicology as well. Many subsequent chapters contain Drug Summary Tables that list, among other properties, the specific adverse effects that can be caused by each drug. This chapter focuses on the mechanisms underlying these adverse effects.
As a general matter, adverse effects range from those that are common and relatively benign to those that pose serious risk of organ damage or death. Even the former group of adverse effects, however, can cause considerable discomfort and lead patients to avoid or reduce their use of medication. Also, in general, the type and risk of adverse effects depend on the margin of safety between the dose required for efficacy and the dose that causes adverse effects. When the margin of safety is large, toxicity results primarily from overdoses; when this margin is small or nonexistent, adverse effects may be manifest at otherwise therapeutic doses. These principles apply both to prescription medications and to over-the-counter drugs such as acetaminophen and aspirin. Note that safety margins are a function not only of the drug but also of the patient, in that genetic or other characteristics—such as polymorphisms in enzymes that detoxify harmful metabolites, comorbidities, or reduced functional reserve in key organs—render patients more or less capable of defending against toxicity. This is one reason why, all other things being equal, new medications should be initiated at the lowest doses likely to be therapeutic.
Drug toxicity is critically important in drug development (see Chapter 51, Drug Discovery and Preclinical Development, and Chapter 52, Clinical Drug Evaluation and Regulatory Approval). Early in drug development, preclinical and clinical studies are used to evaluate compound potency, selectivity, pharmacokinetic and metabolic profiles, and toxicity. Prior to marketing, the regulatory agencies responsible for drug approval review the test data and decide whether the benefits of the drug outweigh its risks. Once a drug is marketed and many more patients are exposed, the appearance of unexpected types or frequencies of adverse effects may cause a reevaluation of the drug, such that its use may be restricted to specific patient populations or withdrawn entirely (as in the cases, for example, of the nonsteroidal anti-inflammatory drug rofecoxib and the antidiabetic drug troglitazone).
In this chapter, categories of drug toxicities that derive from inappropriate activation or inhibition of the intended drug target (on-target adverse effects) or unintended targets (off-target adverse effects) are discussed first. The phenotypic effects of these drug toxicities are then discussed at the physiologic, cellular, and molecular levels. General principles and specific examples are also illustrated in this chapter and throughout the book. The development of rational therapeutic strategies often requires an understanding of the mechanisms of both drug action and drug toxicity.
 Ms. G is an 80-year-old piano teacher with progressively severe right leg pain over a period of 5 to 10 years. She has continued to teach in her studio but at the cost of increasing pain and fatigue. Imaging studies reveal severe osteoarthritis of the right hip. She is scheduled for elective replacement of the right hip with a prosthetic joint.
Ms. G is an 80-year-old piano teacher with progressively severe right leg pain over a period of 5 to 10 years. She has continued to teach in her studio but at the cost of increasing pain and fatigue. Imaging studies reveal severe osteoarthritis of the right hip. She is scheduled for elective replacement of the right hip with a prosthetic joint.
The total hip replacement is performed without immediate complications. During the first few days after the operation, Ms. G is given low-molecular-weight heparin and warfarin as prophylaxis against deep vein thrombosis. Six days after the operation, she develops excruciating pain in the area of the operation. Right lateral hip and buttock swelling is noted on physical examination. A complete blood count reveals significant blood loss (drop in hematocrit from 35% to 25%), and she is taken back to the operating room for evacuation of a large hematoma around the prosthetic joint. Although the hematoma does not appear to be grossly infected, cultures of the hematoma are positive for Staphylococcus aureus.
Because prosthetic joint infections are difficult to treat successfully without removal of the prosthesis, Ms. G is started on an aggressive 12-week course of combination antibiotics in which intravenous vancomycin and oral rifampin are administered for 2 weeks followed by oral ciprofloxacin and rifampin for 10 weeks. She tolerates the first 2 weeks of antibiotics without complications. However, 36 hours after switching her antibiotic from vancomycin to ciprofloxacin, she develops a high fever to 103°F and extreme weakness. Aspiration of the hip reveals only a scant amount of straw-colored (i.e., nonpurulent) fluid. Ms. G is therefore admitted to the hospital for close observation.
Twelve hours after her admission, Ms. G develops an extensive maculopapular rash over her chest, back, and extremities. Her ciprofloxacin and rifampin are discontinued, and vancomycin is restarted. Gradually, over the next 72 hours, her temperature returns to normal and her rash begins to fade. There is no growth in the culture of the right hip aspirate. Ms. G is continued on vancomycin as a single agent for the next 4 weeks without incident; rifampin is restarted, again without incident; and the 12-week antibiotic course is eventually completed using a combination of trimethoprim-sulfamethoxazole and rifampin.
Four months after her hip surgery, Ms. G is back to teaching her piano students and making slow but steady progress in her rehabilitation program.
Questions
1. How likely was it that Ms. G’s high fever, weakness, and skin rash represented a drug reaction to ciprofloxacin?
2. What was the rationale for co-administration of low-molecular-weight heparin and warfarin in the immediate postoperative period?
3. Was there a cause-and-effect relationship between administration of the prophylactic anticoagulants and Ms. G’s life-threatening bleeding complication?

Whether a drug will do more harm than good in an individual patient depends on many factors, including the patient’s age, genetic makeup, and preexisting conditions; the dose of the drug administered; and other drugs that the patient may be taking. For example, the very old or very young may be more susceptible to the toxic effects of a drug because of age-dependent differences in pharmacokinetic profiles or drug-metabolizing enzymes. As discussed in Chapter 4, Drug Metabolism, genetic factors may determine individual characteristics of drug metabolism, receptor activity, or repair mechanisms. Adverse drug reactions may be more likely in patients with preexisting conditions, such as liver or kidney dysfunction, and, of course, in patients allergic to specific drugs. Concomitant medications can confound both efficacy and toxicity of drugs, particularly when these medications share or modulate the same metabolic pathways or transporters. Drug interactions with health supplements are also an important but often under-recognized cause of drug toxicity. Drug–drug and drug–herb interactions are discussed later in this chapter. The clinical determination of a drug’s toxicity may not always be straightforward: as in the case of Ms. G, for example, a patient being treated with an antibiotic to combat an infection can develop a high fever, skin rash, and significant morbidity due either to recurrence of the infection or, instead, to an adverse reaction to the antibiotic.
Although a spectrum of adverse effects may be associated with the use of any drug or drug class, it is helpful to conceptualize the mechanisms of drug toxicity based on several general paradigms:
 “On-target” adverse effects, which are the result of the drug binding to its intended receptor, but at an inappropriate concentration, with suboptimal kinetics, or in the incorrect tissue (Fig. 6-1)
“On-target” adverse effects, which are the result of the drug binding to its intended receptor, but at an inappropriate concentration, with suboptimal kinetics, or in the incorrect tissue (Fig. 6-1)
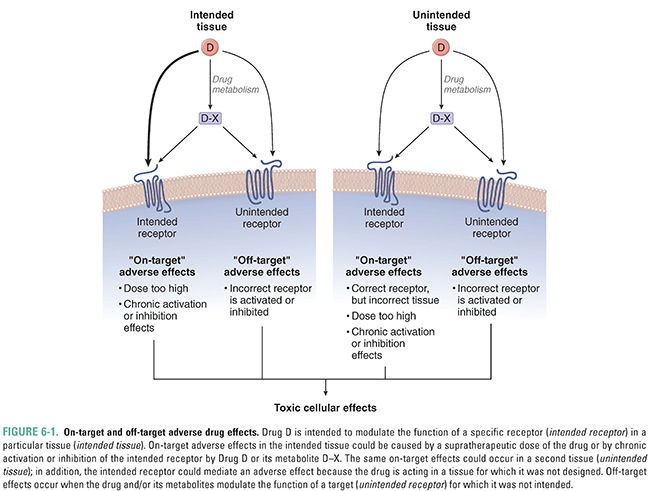
 “Off-target” adverse effects, which are caused by the drug binding to a target or receptor for which it was not intended (Fig. 6-1)
“Off-target” adverse effects, which are caused by the drug binding to a target or receptor for which it was not intended (Fig. 6-1)
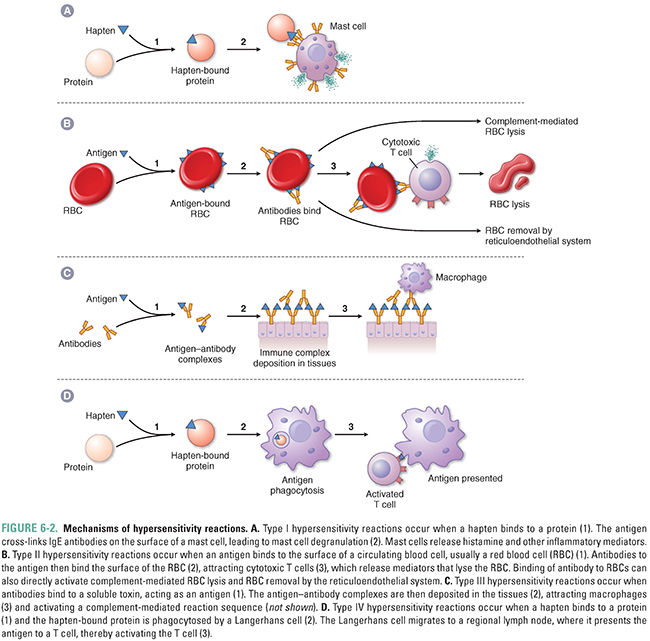
 Adverse effects mediated by the immune system (Fig. 6-2)
Adverse effects mediated by the immune system (Fig. 6-2)
 Idiosyncratic responses for which the mechanism is not known
Idiosyncratic responses for which the mechanism is not known
These four mechanisms are discussed below. Note that many drugs can have both on-target and off-target effects, and adverse effects observed in patients can be due to multiple mechanisms.
An important concept in drug toxicity is that an adverse effect may be an exaggeration of the desired pharmacologic action as a result of changes in exposure or sensitivity to the drug (see Fig. 6-1). This can occur due to deliberate or accidental overdoses, alterations in the pharmacokinetics of the drug (e.g., due to liver or kidney disease or to interactions with other drugs), or changes in the pharmacodynamics of the drug–receptor interaction that alter the pharmacologic response (e.g., an increase in receptor number). All such changes can lead to an increase in the effective concentration of the drug and thus to an increased biological response. Because on-target effects are mediated via the desired mechanism of action of the drug, these effects are often shared by every member of the therapeutic class and are thus also known as class effects.
An important set of on-target adverse effects may occur because the drug, or one of its metabolites, interacts with the appropriate receptor but in tissues other than those affected by the disease condition being treated (Fig. 6-1). Many drug targets are expressed in more than one cell type or tissue. For example, the antihistamine diphenhydramine is an H1 receptor antagonist used to ameliorate the effects of histamine release in allergic conditions. This drug also crosses the blood–brain barrier to antagonize H1 receptors in the central nervous system, leading to somnolence. This adverse effect led to the design of second-generation H1 receptor antagonists that do not cross the blood–brain barrier and thus do not induce drowsiness. Notably, the first of these second-generation H1 antagonists, terfenadine, produced an off-target effect (interaction with cardiac potassium channels) that led to a different and serious adverse effect—an increased risk of cardiac death. This example is discussed later in this chapter.
Local anesthetics such as lidocaine and bupivacaine provide a second example of an on-target adverse effect. These drugs are intended to prevent axonal impulse transmission by blocking sodium channels in neuronal membranes near the site of injection. Blockade of sodium channels in the central nervous system (CNS) following overdose or inappropriate administration (e.g., intravascular administration) can lead to tremors, seizures, and death. These on-target effects are discussed in greater detail in Chapter 12, Local Anesthetic Pharmacology.
The antipsychotic agent haloperidol is thought to produce its beneficial effect through blockade of mesolimbic and mesocortical D2 receptors. One consequence of blocking D2 receptors in the pituitary gland is an increase in prolactin secretion, leading in some cases to amenorrhea, galactorrhea, sexual dysfunction, and osteoporosis. These on-target effects are discussed in Chapter 14, Pharmacology of Dopaminergic Neurotransmission.
Sometimes, on-target adverse effects unmask important functions of the biological target. A prominent example of this phenomenon occurs with administration of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (so-called statins), which are used to decrease cholesterol levels. The intended target tissue of these drugs is the liver, where they inhibit HMG-CoA reductase, the rate-limiting enzyme of isoprenoid synthesis. A rare adverse effect of statin therapy is muscle toxicity, including rhabdomyolysis and myositis; the fact that this effect occurs highlights the physiologic role of HMG-CoA reductase in regulating the post-translational modification of several muscle proteins through a lipidation process called geranyl-geranylation. Statins, as examples of drugs causing skeletal muscle injury, are also referenced later in this chapter.
Very few drugs are so selective that they interact with only one molecular target. Off-target adverse effects occur when a drug interacts with unintended targets (Fig. 6-1). A prominent example of an off-target effect is the interaction of numerous compounds with cardiac IKr potassium channels. (Because the human ether-à-go-go-related gene [hERG] codes for one subunit of the human IKr channel, these channels are also called hERG channels.) Inhibition of potassium currents carried by IKr channels can lead to delayed repolarization of cardiac myocytes (see Chapter 24, Pharmacology of Cardiac Rhythm). In turn, delayed repolarization can lead to an increase in the heart-rate corrected QT interval (QTc), cardiac arrhythmias including torsades de pointes, and sudden death. The antihistamine terfenadine was one of the earliest examples of compounds found to interfere with cardiac potassium channel currents, leading to potentially fatal arrhythmias. This drug was designed to avoid drowsiness, an adverse effect of the first-generation H1 antagonists (see earlier discussion). The observation of increased deaths due to cardiac arrhythmias in patients receiving terfenadine led to both withdrawal of this compound from the market and vigorous efforts to understand how to prevent such events. It is now known that, although many compounds inhibit the hERG channel, compounds with a half-maximal inhibitory concentration (IC50) more than 30-fold greater than the maximum plasma concentration at the recommended therapeutic dose (Cmax, adjusted for protein binding) pose a low risk of causing QTc prolongation and cardiac arrhythmia. The active metabolite of terfenadine, fexofenadine, inhibits the hERG channel only weakly, and fexofenadine is now marketed as a safer antihistamine.
Many compounds can interfere with cardiac potassium channels: accordingly, all new drug candidates are evaluated for the potential to interact with these promiscuous channels. In the hERG assay, the potential effect of compounds on human cardiac potassium currents is measured in an in vitro system using cells transfected with hERG. In addition to the hERG assay, the potential for altering cardiac electrophysiology is evaluated in a nonrodent animal model (see Chapter 51). As a condition of marketing approval, new drugs are also evaluated for their ability to prolong QTc in humans; this evaluation is generally performed in large, late-stage clinical trials. Compounds that increase QTc by more than a specified value at an exposure near that required for the therapeutic effect are considered to pose a risk for producing arrhythmias. The positive control compound used in many of these “thorough QTc studies” is moxifloxacin, an antibiotic that increases QTc at clinical doses (but confers a low risk of arrhythmogenesis). While the approach of relying most heavily on hERG inhibition in vitro and on thorough QTc clinical trials has prevented the recent introduction of “torsadogenic” drugs, this approach has been criticized for its low sensitivity, cost, and lack of assessment of the impact of other ion channel effects that mitigate or enhance the effects of compounds on QTc. Based on these concerns, alternative approaches are being considered, including preclinical evaluations that study a broader range of ion channel effects and clinical studies at an earlier stage of drug development.
Enantiomers (mirror-image isomers) of a drug can also cause off-target effects. As described in Chapter 1, Drug–Receptor Interactions, drug receptors are exquisitely sensitive to the three-dimensional structure of the drug molecule; therefore, receptors can often distinguish between enantiomers of a drug. A tragic and well-known example of this phenomenon occurred with the administration of racemic thalidomide (mixture of [R]- and [S]-enantiomers) in the 1960s as a treatment for morning sickness in pregnant women. While the [R]-enantiomer of thalidomide was an effective sedative, the [S]-enantiomer was a potent teratogen that caused severe birth defects such as amelia (absence of limbs) and various degrees of phocomelia in an estimated 10,000 newborns in 46 countries (but not in the United States, thanks to Frances Kelsey at the Food and Drug Administration [FDA], who doubted the safety of thalidomide and prevented its approval). The use of drugs in pregnant patients is discussed later in this chapter (see “Teratogenesis Due to Drug Therapy” and Box 6-1).
BOX 6-1 Application to Therapeutic Decision Making: Drugs in Pregnancy |
Prescribing drugs to women who are, or might become, pregnant requires a risk–benefit evaluation for both the mother and the fetus. However, many drugs have not been systematically studied in pregnant populations, so such risk–benefit evaluations may be uncertain. The FDA places drugs into five “pregnancy categories” based on data from studies in laboratory animals, observations from well-controlled epidemiologic studies (or lack thereof), and/or case reports. These categories appear on drug labels and are listed below. Note that the categories are not strictly scaled according to risk; although Category A drugs are typically the safest for use in pregnancy, and Category X drugs are, as the name suggests, contraindicated, drugs in Category B—for which, by definition, human data are limited or inadequate—are not necessarily “almost as safe” as those in Category A. Category A Adequate and well-controlled studies have failed to demonstrate a risk to the fetus in the first trimester of pregnancy (and there is no evidence of risk in later trimesters). Category B Animal reproduction studies have failed to demonstrate a risk to the fetus, and there are no adequate and well-controlled studies in pregnant women. Category C Animal reproduction studies have shown an adverse effect on the fetus, and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks. Category D There is positive evidence of human fetal risk based on adverse reaction data from investigational or marketing experience or studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks. Category X Studies in animals or humans have demonstrated fetal abnormalities and/or there is positive evidence of human fetal risk based on adverse reaction data from investigational or marketing experience, and the risks involved in use of the drug in pregnant women clearly outweigh potential benefits. Category X drugs include not only teratogens but also drugs that have no proper use in pregnant patients. Statins are in this category, for example, because the normal physiologic increase in serum cholesterol that occurs during pregnancy should not be suppressed. Despite their long history of use, the label categories remain a source of confusion, even occasionally within the FDA itself. For example, the antibiotic tigecycline is classified in Category D, but the absence of controlled data from humans indicates that it should instead be placed in Category C. More generally, the FDA pregnancy categorization of drugs, like any such scheme, is not perfect and may fail to capture the nuances of some drug-specific and patient-specific circumstances. Thus, the physician should also rely on his or her own judgment, keeping in mind the following issues:
When appropriate, drugs that have proven effective for treating a patient’s condition should be continued. To minimize fetal risk, drugs should be prescribed at the lowest therapeutic dose, taking into account the metabolic and physiologic changes that occur during pregnancy. |




The potential for significant pharmacologic differences between drug enantiomers has led researchers and the FDA to evaluate such compounds as separate chemical entities. If a single enantiomeric preparation of a drug can be shown to have improved pharmacologic properties over a racemic version, then the purified enantiomer can be approved as a new drug. For example, the racemic proton pump inhibitor omeprazole and its [S]-enantiomer esomeprazole (as in [S]-omeprazole) are marketed as separate drugs.
Another common off-target effect is the unintended activation of different receptor subtypes. For example, the β1-adrenergic receptor is expressed in the heart, and its activation increases heart rate and myocardial contractility. Closely related β2-adrenergic receptors are expressed in smooth muscle cells in the airways and in the vasculature, and activation of β2 receptors leads to smooth muscle relaxation and dilation of these tissues (see Chapter 11, Adrenergic Pharmacology). The clinical uses of β-adrenergic receptor antagonists (β-blockers) are often targeted to the β1 receptor to control heart rate and reduce myocardial oxygen demand in patients with angina or heart failure. However, some β1 receptor antagonists are not entirely selective for the β1 receptor and can also antagonize the β2 receptor. β-Adrenergic receptor antagonists with nonselective effects are therefore contraindicated in patients with asthma, because such drugs could cause bronchoconstriction by antagonizing β2 receptors. Similarly, the use of inhaled β2 agonists in the treatment of asthma, particularly at high doses, may lead to increased heart rate.
A second off-target effect due to unintended activation of different receptor subtypes is the valvulopathy caused by the anorectic agent fenfluramine. This drug’s primary mechanism of action appears to involve release of serotonin (5-hydroxytryptamine [5-HT]) and inhibition of 5-HT reuptake in brain areas that regulate feeding behavior. However, the compound also activates 5-HT2B receptors, leading to proliferation of myofibroblasts in the atrioventricular valves. Pulmonary hypertension can develop and, in some cases, lead to death. Because of this adverse effect, fenfluramine has been withdrawn from the market (see “Drug-Induced Cardiovascular Toxicity”).
The potential off-target effects of some drugs can be explored by using genetically modified laboratory mice or rats in which the intended target receptor has been deleted (sometimes only in specific tissues). If the drug nonetheless affects the physiology of these rodents, then targets other than the intended target must be involved.
Off-target effects of some drugs and drug metabolites can be determined only empirically, underscoring the importance of extensive drug testing both in preclinical experiments and in clinical trials. Despite such testing, some rare drug toxicities are discovered only when exposure occurs in a much larger population than that required for clinical trials. For example, fluoroquinolones, a class of broad-spectrum antibiotics derived from nalidixic acid, displayed minimal toxicities in preclinical studies and clinical trials. Wider clinical use of these drugs, however, led to reports of anaphylaxis, QTc prolongation, and potential cardiotoxicity, resulting in the removal of two drugs of this class, temafloxacin and grepafloxacin, from the market. Use of another fluoroquinolone, trovafloxacin, is significantly restricted due to hepatic toxicity. In comparison, ciprofloxacin and levofloxacin are generally well tolerated and are frequently used in the treatment of bacterial infections. As seen in the introductory case, however, even these agents can occasionally cause a severe drug hypersensitivity reaction.
Idiosyncratic drug reactions are adverse effects that appear unpredictably, in a small fraction of patients, for unknown reasons. These effects are not typically manifest in premarketing testing in either laboratory animals or patients. The appearance of idiosyncratic injury leading to permanent organ dysfunction and/or death, even if rare, often prompts withdrawal of the drug from the market, precisely because susceptible patient populations cannot be identified. The systematic study of patient variations in response to different drugs may help to elucidate the genetic or other mechanisms that underlie idiosyncratic drug reactions.

The Swiss physician and alchemist Paracelsus noted nearly 500 years ago that “all substances are poison; there is none which is not a poison. The right dose differentiates a poison and a remedy.” In some cases, such as a suicide or homicide, the overdose of a drug is intentional. Most cases of overdose, however, are accidents. Adverse drug events due to medication errors are estimated to affect some 7 million people each year, with associated costs of $21 billion annually. This significant cost to both the patient and the health care system has led to systematic efforts intended to minimize errors in prescribing and dosing practices.
As the population has aged and increasing numbers of patients have been prescribed multiple medications, the potential for drug–drug interactions has grown. Numerous adverse interactions have been identified, and the mechanisms often involve pharmacokinetic or pharmacodynamic effects. Drug–herb interactions are also an important subset of drug–drug interactions.
Pharmacokinetic Drug–Drug Interactions
Pharmacokinetic interactions arise when one drug changes the absorption, distribution, metabolism, or excretion of another drug, thereby altering the concentration of active drug in the body. As discussed in Chapter 4, some drugs can inhibit or induce hepatic P450 enzymes. If two drugs are metabolized by the same P450 enzyme, competitive or irreversible inhibition of that P450 enzyme by one drug can result in an increase in the plasma concentration of the second drug. On the other hand, induction of a specific P450 enzyme by one drug can lead to a decrease in the plasma concentrations of other drugs that are metabolized by the same enzyme. The antifungal drug ketoconazole is a potent inhibitor of cytochrome P450 3A4 (CYP3A4). Co-administration of drugs that are also metabolized by CYP3A4 may result in reduced metabolism of these drugs and higher plasma drug levels. If the co-administered drug has a low therapeutic index, toxicity may occur. Because of its potent inhibition of CYP3A4, ketoconazole is often used in clinical studies designed to assess the importance of pharmacokinetic drug–drug interactions.
In addition to altering the activity of P450 enzymes, drugs can affect the transport of other drugs into and out of tissues (see Chapter 4 and Chapter 5, Drug Transporters). For example, P-glycoprotein (P-gp), encoded by the multidrug resistance 1 (MDR1) gene, is an efflux pump that transports drugs into the intestinal lumen. Administration of a drug that inhibits or is a substrate for P-gp can lead to an increase in the plasma concentration of other drugs that are normally pumped out of the body by this mechanism. Since P-gp also plays a role in transport of drugs across the blood–brain barrier, compounds that inhibit P-gp can affect drug transport into the CNS. Other transporters, such as the organic anion transporting polypeptide 1 (OATP1), mediate uptake of drugs into hepatocytes for metabolism and transport of drugs across the tubular epithelium of the kidney for excretion; both of these mechanisms promote clearance of drugs from the body. Interactions of a drug or one of its metabolites with these classes of transporters can lead to inappropriately high plasma concentrations of other drugs that are handled by the same transporter.
A pharmacokinetic interaction can sometimes be desirable. For example, because penicillin is cleared via tubular secretion in the kidney, the elimination half-life of this drug can be increased if the drug is given concomitantly with probenecid, an inhibitor of renal tubular transport. A second example is the combination of imipenem, a broad-spectrum antibiotic, with cilastatin, a selective inhibitor of a renal brush border dipeptidase (dehydropeptidase I). Because imipenem is rapidly inactivated by dehydropeptidase I, co-administration of imipenem with cilastatin is used to achieve therapeutic plasma concentrations of the antibiotic.
A drug that binds to plasma proteins (such as albumin) may displace a second drug from the same proteins to increase its free plasma concentration and thereby increase its bioavailability to target and nontarget tissues. This effect can be enhanced in a situation in which circulating albumin levels are low, such as liver failure or malnutrition (decreased albumin synthesis) or nephrotic syndrome (increased albumin excretion).
Pharmacodynamic Drug–Drug Interactions
Pharmacodynamic interactions arise when one drug changes the response of target or nontarget tissues to another drug. Toxic pharmacodynamic interactions can occur when two drugs activate complementary pathways, leading to an exaggerated biological effect. Such a drug interaction occurs upon co-administration of sildenafil (for erectile dysfunction) and nitroglycerin (for angina pectoris). Sildenafil inhibits phosphodiesterase type 5 (PDE5) and thus prolongs the action of cyclic GMP (cGMP), and nitroglycerin stimulates guanylyl cyclase to increase cGMP levels in vascular smooth muscle. Co-exposure to the two drugs increases cGMP to an even greater degree, increasing the risk of severe hypotension (see Chapter 22, Pharmacology of Vascular Tone).
A second example is the co-administration of antithrombotic drugs. After hip replacement surgery, patients are often treated with prophylactic warfarin for a number of weeks to prevent the development of postoperative deep vein thrombosis. Because plasma warfarin concentrations may not reach a therapeutic level for several days, patients are sometimes co-administered low-molecular-weight heparin and warfarin during this time. As seen in the case of Ms. G, however, significant bleeding may result if the effects of the heparin and warfarin synergize to produce supratherapeutic levels of anticoagulation. Newer antithrombotic drugs used for prophylaxis after orthopedic surgery (e.g., apixaban, rivaroxaban) may avoid this increased risk of bleeding because they reach therapeutic concentrations rapidly and do not require co-administration with another antithrombotic drug such as heparin.
The safety and efficacy of a drug may also be altered by co-exposure with various nonpharmaceuticals, such as foods, beverages, and herbal and other dietary supplements. Many herbal products are complex mixtures of biologically active compounds, and their safety and effectiveness have rarely been tested in controlled studies. The wide use of unregulated herbal products among the public should lead clinicians to inquire about patient use of such products.
The literature contains some reports of therapeutic failure of drugs taken in conjunction with herbal products and some reports of toxicity. For example, the herbal preparation ginkgo biloba (from the tree of the same name) inhibits platelet aggregation. Concomitant use of ginkgo and nonsteroidal anti-inflammatory drugs (NSAIDs), which also inhibit platelet aggregation, may increase the risk of bleeding. In combination with selective serotonin reuptake inhibitors, St. John’s wort may cause serotonin syndrome.
Cellular Mechanisms of Toxicity: Apoptosis and Necrosis
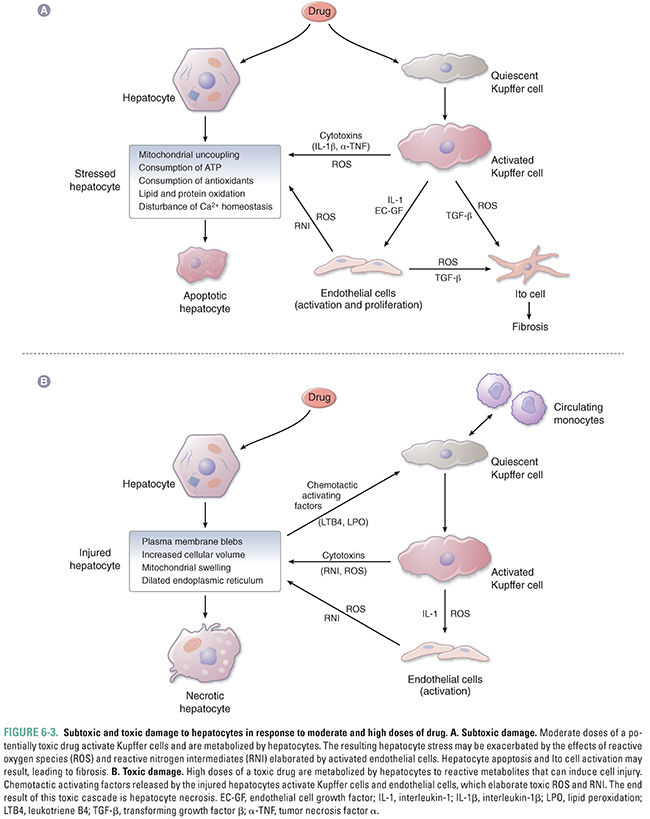
Cells are equipped with various mechanisms to avoid or repair damage: toxicity occurs if and when these defenses are overwhelmed. In some cases, toxicity can be minimized in the short term, but repeated insults (e.g., those leading to fibrosis) can eventually compromise organ function.
The primary cellular responses to a potentially toxic drug are illustrated in Figure 6-3A and 6-3B, using the hepatocyte as an example. Depending on the severity of the toxic insult, a cell may undergo apoptosis (programmed cell death) or necrosis (uncontrolled cell death). Apoptosis allows the cell to undergo ordered self-destruction by the coordinated activation of a number of dedicated proteins. Apoptosis can be beneficial when it eliminates damaged cells without damage to surrounding tissue. Inhibition of apoptosis is common in many cancer cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


