Francis J. Alenghat and David E. Golan
PROCESSING OF SIGNALS RESULTING FROM DRUG–RECEPTOR INTERACTIONS |

Why is it that one drug affects cardiac function and another alters the transport of specific ions in the kidney? Why do antibiotics effectively kill bacteria but rarely harm patients? These questions can be answered by first examining the interaction between a drug and its specific molecular target and then considering the role of that action in a broader physiologic context. This chapter focuses on the molecular details of drug–receptor interactions, emphasizing the variety of receptors and their molecular mechanisms. This discussion provides a conceptual basis for the action of the many drugs and drug classes discussed in this book. It also serves as a background for Chapter 2, Pharmacodynamics, which discusses the quantitative relationships between drug–receptor interactions and pharmacologic effect.
Although drugs can theoretically bind to almost any three-dimensional target, most drugs achieve their desired (therapeutic) effects by interacting selectively with target molecules that play important physiologic or pathophysiologic roles. In many cases, selectivity of drug binding to receptors also determines the undesired (adverse) effects of a drug. In general, drugs are molecules that interact with specific molecular components of an organism to cause biochemical and physiologic changes within that organism. Drug receptors are macromolecules that, upon binding to a drug, mediate those biochemical and physiologic changes.
 Intent on enjoying his newly found retirement, Mr. B has made a point of playing tennis as often as possible during the past year. For the past 3 months, however, he has noted increasing fatigue. Moreover, he is now unable to finish a meal, despite his typically voracious appetite. Worried and wondering what these symptoms mean, Mr. B schedules an appointment with his doctor. On physical examination, the physician notes that Mr. B has an enlarged spleen, extending approximately 10 cm below the left costal margin; the physical exam is otherwise within normal limits. Blood tests show an increased total white blood cell count (70,000 cells/mm3) with an absolute increase in neutrophils, band forms, metamyelocytes, and myelocytes, but no blast cells (undifferentiated precursor cells). Cytogenetic analysis of metaphase cells demonstrates that 90% of Mr. B’s myeloid cells possess the Philadelphia chromosome (indicating a translocation between chromosomes 9 and 22), confirming the diagnosis of chronic myeloid leukemia. The physician initiates therapy with imatinib, a highly selective inhibitor of the BCR-Abl tyrosine kinase fusion protein that is encoded by the Philadelphia chromosome. Over the next month, the cells containing the Philadelphia chromosome disappear completely from Mr. B’s blood, and he begins to feel well enough to compete in a seniors tennis tournament. Mr. B continues to take imatinib every day, and he has a completely normal blood count and no fatigue. He is not sure what the future will bring, but he is glad to have been given the chance to enjoy a healthy retirement.
Intent on enjoying his newly found retirement, Mr. B has made a point of playing tennis as often as possible during the past year. For the past 3 months, however, he has noted increasing fatigue. Moreover, he is now unable to finish a meal, despite his typically voracious appetite. Worried and wondering what these symptoms mean, Mr. B schedules an appointment with his doctor. On physical examination, the physician notes that Mr. B has an enlarged spleen, extending approximately 10 cm below the left costal margin; the physical exam is otherwise within normal limits. Blood tests show an increased total white blood cell count (70,000 cells/mm3) with an absolute increase in neutrophils, band forms, metamyelocytes, and myelocytes, but no blast cells (undifferentiated precursor cells). Cytogenetic analysis of metaphase cells demonstrates that 90% of Mr. B’s myeloid cells possess the Philadelphia chromosome (indicating a translocation between chromosomes 9 and 22), confirming the diagnosis of chronic myeloid leukemia. The physician initiates therapy with imatinib, a highly selective inhibitor of the BCR-Abl tyrosine kinase fusion protein that is encoded by the Philadelphia chromosome. Over the next month, the cells containing the Philadelphia chromosome disappear completely from Mr. B’s blood, and he begins to feel well enough to compete in a seniors tennis tournament. Mr. B continues to take imatinib every day, and he has a completely normal blood count and no fatigue. He is not sure what the future will bring, but he is glad to have been given the chance to enjoy a healthy retirement.
Questions
1. How does imatinib interrupt the activity of the BCR-Abl tyrosine kinase fusion protein?
2. Unlike imatinib, most of the older therapies for chronic myeloid leukemia (such as interferon-α) had significant “flu-like” adverse effects. Why did these therapies cause significant adverse effects in most patients, whereas (as in this case) imatinib causes adverse effects in very few patients?
3. Why is imatinib a selective therapy for chronic myeloid leukemia? Is this selectivity related to the lack of adverse effects associated with imatinib therapy?
4. How does the BCR-Abl protein affect intracellular signaling pathways?
 CONFORMATION AND CHEMISTRY OF DRUGS AND RECEPTORS
CONFORMATION AND CHEMISTRY OF DRUGS AND RECEPTORS
An understanding of why a drug binds to a particular receptor can be found in the structure and chemical properties of the two molecules. This section discusses the basic determinants of receptor structure and the chemistry of drug–receptor binding. The discussion here focuses primarily on the interactions of drugs that are small molecules with target receptors that are mainly macromolecules (especially proteins), but many of these principles also apply to the interactions of antibody- or other protein-based therapeutics with their molecular targets (see Chapter 54, Protein Therapeutics).
Because many human and microbial drug receptors are proteins, it is useful to review the four major levels of protein structure (Fig. 1-1). At the most basic level, proteins consist of long chains of amino acids, the sequences of which are determined by the sequences of the DNA that code for the proteins. A protein’s amino acid sequence is referred to as its primary structure. Once a long chain of amino acids has been synthesized on a ribosome, many of the amino acids begin to interact with nearby amino acids in the polypeptide chain. These interactions, which are typically mediated by hydrogen bonding, give rise to the secondary structure of a protein by forming well-defined conformations such as the α helix, β pleated sheet, and β barrel. As a result of their highly organized shape, these structures often pack tightly with one another, further defining the overall shape of the protein. Tertiary structure results from the interaction of amino acids more distant from one another along a single amino acid chain. These interactions include hydrogen bond and ionic bond formation as well as the covalent linkage of sulfur atoms to form intramolecular disulfide bridges. Finally, polypeptides may oligomerize to form more complex structures. The conformation that results from the interaction of separate polypeptides is referred to as the quaternary structure.
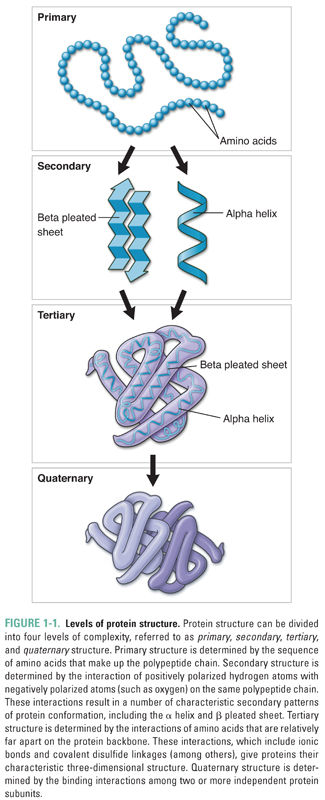
Different portions of a protein’s structure generally have different affinities for water, and this feature has an additional effect on the protein’s shape. Because both the extracellular and intracellular environments are composed primarily of water, hydrophobic protein segments are often drawn to the inside of the protein or shielded from water by insertion into lipid bilayer membranes. Conversely, hydrophilic protein segments are often located on a protein’s exterior surface. After all of this twisting and turning is completed, each protein has a unique shape that determines its function, location in the body, relationship to cellular membranes, and binding interactions with drugs and other macromolecules.
The site on the receptor at which the drug binds is called its binding site. Each binding site has unique chemical characteristics that are determined by the specific properties of the amino acids that make up the site. The three-dimensional structure, shape, and reactivity of the site, and the inherent structure, shape, and reactivity of the drug, determine the orientation of the drug with respect to the receptor and govern how tightly these molecules bind to one another. Drug–receptor binding is the result of multiple chemical interactions between the two molecules, some of which are fairly weak (such as van der Waals forces) and some of which are extremely strong (such as covalent bonding). The sum total of these interactions provides the specificity of the overall drug–receptor interaction. The favorability of a drug–receptor interaction is referred to as the affinity of the drug for its binding site on the receptor. This concept is discussed in more detail in Chapter 2. The chemistry of the local environment in which these interactions occur—such as the hydrophobicity, hydrophilicity, and pKa of amino acids near the binding site—may also affect the affinity of the drug–receptor interaction. The primary forces that contribute to drug–receptor affinity are described below and in Table 1-1.

van der Waals forces, resulting from the polarity induced in a molecule by the shifting of its electron density in response to the close proximity of another molecule, provide a weak attractive force for drugs and their receptors. This induced polarity is a ubiquitous component of all molecular interactions. Hydrogen bonds have substantial strength and are often important for drug–receptor association. This type of bond is mediated by the interaction between positively polarized hydrogen atoms (which are covalently attached to more electronegative atoms such as nitrogen or oxygen) and negatively polarized atoms (such as oxygen, nitrogen, or sulfur that are covalently attached to less electronegative atoms such as carbon or hydrogen). Ionic interactions, which occur between atoms with opposite charges, are stronger than hydrogen bonds but less strong than covalent bonds. Covalent bonding results from the sharing of a pair of electrons between two atoms on different molecules. Covalent interactions are so strong that, in most cases, they are essentially irreversible. Table 1-1 indicates the mechanism of interaction and relative strength of each of these types of bonds. As noted above, the environment in which drugs and receptors interact also affects the favorability of binding. The hydrophobic effect refers to the mechanism by which the unique properties of the ubiquitous solvent water cause the interaction of a hydrophobic molecule with a hydrophobic binding site to be enhanced.
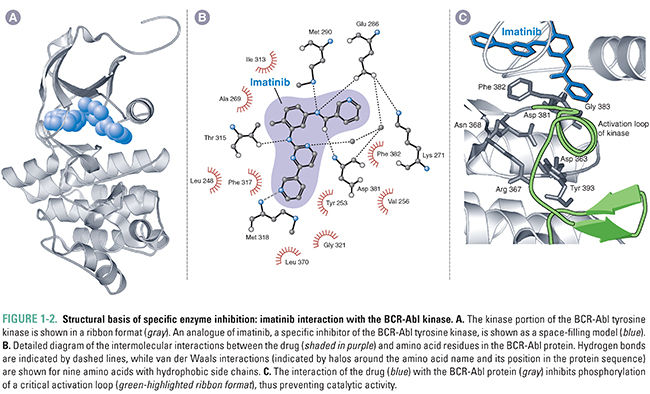
Rarely is drug–receptor binding caused by a single type of interaction; rather, it is a combination of these binding interactions that provides drugs and receptors with the forces necessary to form a stable drug–receptor complex. In general, multiple weak forces comprise the majority of drug–receptor interactions. For example, imatinib forms many van der Waals interactions and hydrogen bonds with the ATP-binding site of the BCR-Abl tyrosine kinase. The sum total of these relatively weak forces creates a strong (high affinity) interaction between this drug and its receptor (Fig. 1-2). Ionic and hydrophobic interactions exert force at a greater distance than van der Waals interactions and hydrogen bonds; for this reason, the former interactions are often critical to initiate the association of a drug and receptor.
Although relatively rare, covalent interactions between a drug and its receptor are a special case. The formation of a covalent bond is often essentially irreversible, and in such cases, the drug and receptor form an inactive complex. To regain activity, the cell must synthesize a new receptor molecule to replace the inactivated protein; and the drug molecule, which is also part of the inactive complex, is generally not available to inhibit other receptor molecules. Drugs that modify their target receptors (often enzymes) through this mechanism are sometimes called suicide substrates. Aspirin is an example of such a drug; it irreversibly acetylates cyclooxygenases to reduce the production of prostaglandins (anti-inflammatory effect) and thromboxanes (antiplatelet effect) (see Chapter 43, Pharmacology of Eicosanoids).
The molecular structure of a drug dictates the physical and chemical properties that contribute to its specific binding to the receptor. Important factors include hydrophobicity, ionization state (pKa), conformation, and stereochemistry of the drug molecule. All of these factors combine to determine the complementarity of the drug to the binding site. Receptor binding pockets are highly specific, and small changes in the drug can have a large effect on the affinity of the drug–receptor interaction. For example, the stereochemistry of the drug has a great impact on the strength of the binding interaction. Warfarin is synthesized and administered as a racemic mixture (a mixture containing 50% of the right-handed molecule and 50% of the left-handed molecule); however, the S enantiomer is four times more potent than the R because of a stronger interaction of the S form with its binding site on vitamin K epoxide reductase. Stereochemistry can also affect toxicity in cases where one enantiomer of a drug causes the desired therapeutic effect and the other enantiomer causes an undesired toxic effect, perhaps due to an interaction with a second receptor or to metabolism to a toxic species. Although it is sometimes difficult for pharmaceutical companies to synthesize and purify individual enantiomers on a large scale, a number of currently marketed drugs are produced as individual enantiomers in cases where one enantiomer has higher efficacy and/or lower toxicity than its mirror image.
Impact of Drug Binding on the Receptor
How does drug binding produce a biochemical and/or physiologic change in the organism? In the case of receptors with enzymatic activity, the binding site of the drug is often the active site at which an enzymatic transformation is catalyzed, and the catalytic activity of the enzyme is inhibited by drugs that prevent substrate binding to the site or that covalently modify the site. In cases where the binding site is not the active site of the enzyme, drugs can cause a change by preventing the binding of endogenous ligands to their receptor binding pockets. In many drug–receptor interactions, however, the binding of a drug to its receptor results in a change in the conformation of the receptor. Altering the shape of the receptor can affect its function, including enhancing the affinity of the drug for the receptor. Such an interaction is often referred to as induced fit, because the receptor’s conformation changes so as to improve the quality of the binding interaction.
The principle of induced fit suggests that drug–receptor binding can have profound effects on the conformation of the receptor. By inducing conformational changes in the receptor, many drugs not only improve the quality of the binding interaction but also alter the action of the receptor. The change in shape induced by the drug is sometimes identical to that caused by the binding of an endogenous ligand. For example, exogenously administered insulin analogues all stimulate the insulin receptor to the same extent, despite their slightly different amino acid sequences. In other cases, drug binding alters the shape of the receptor so as to make it more or less functional than normal. For example, imatinib binding to the BCR-Abl tyrosine kinase causes the protein to assume an enzymatically inactive conformation, thus inhibiting the kinase activity of the receptor.
Another way to describe the induced fit principle is to consider that many receptors exist in multiple conformational states—such as inactive (or closed), active (or open), and desensitized (or inactivated)—and that the binding of a drug to the receptor stabilizes one or more of these conformations. Quantitative models that incorporate these concepts of drug–receptor interactions are discussed in Chapter 2.
Membrane Effects on Drug–Receptor Interactions
The structure of the receptor also determines where the protein is located in relationship to cellular boundaries such as the plasma membrane. Proteins that have large hydrophobic domains are able to reside in the plasma membrane because of the membrane’s high lipid content. Many receptors that span the plasma membrane have lipophilic domains that are located in the membrane and hydrophilic domains that reside in the intracellular and extracellular spaces. Other drug receptors, including a number of transcription regulators (also called transcription factors), have only hydrophilic domains and reside in the cytoplasm, nucleus, or both.
Just as the structure of the receptor determines its location in relationship to the plasma membrane, the structure of a drug affects its ability to gain access to the receptor. For example, many drugs that are highly water-soluble are unable to pass through the plasma membrane and bind to target molecules in the cytoplasm. Certain hydrophilic drugs are able to pass through transmembrane channels (or use other transport mechanisms) and gain ready access to cytoplasmic receptors. Drugs that are highly lipophilic, such as many steroid hormones, are often able to pass through the hydrophobic lipid environment of the plasma membrane without special channels or transporters and thereby gain access to intracellular targets.
Drug-induced alterations in receptor shape can allow drugs bound to cell surface receptors to affect functions inside cells. Many cell surface receptors have extracellular domains that are linked to intracellular effector molecules by receptor domains that span the plasma membrane and extend into the cytoplasm. In some cases, changing the shape of the extracellular domain can alter the conformation of the membrane-spanning and/or intracellular domains of the receptor, resulting in a change in receptor function. In other cases, drugs can cross-link the extracellular domains of two receptor molecules, forming a dimeric receptor complex that activates effector molecules inside the cell.
All of these factors—drug and receptor structure, the chemical forces influencing drug–receptor interaction, drug solubility in water and in the plasma membrane, and the function of the receptor in its cellular environment—confer substantial specificity on the interactions between drugs and their target receptors. This book discusses numerous examples of drugs that can gain access and bind to receptors, induce conformational changes in the receptors, and thereby produce biochemical and physiologic effects. Specificity of drug–receptor binding suggests that, armed with the knowledge of the structure of a receptor, one could theoretically design a drug that interrupts or enhances receptor activity. This process, known as rational drug design, could potentially increase the efficacy and reduce the toxicity of drugs by optimizing their structure so that they bind more selectively to their targets. Rational drug design was first used to develop highly selective agents such as the antiviral protease inhibitor ritonavir and the antineoplastic tyrosine kinase inhibitor imatinib. Indeed, further rounds of rational drug design have led to the development of second-generation protease inhibitors and antineoplastics with high affinity for the mutated drug targets that can evolve in patients who develop resistance to first-generation drugs. The rational drug design approach is discussed in greater detail in Chapter 51, Drug Discovery and Preclinical Development.
 MOLECULAR AND CELLULAR DETERMINANTS OF DRUG SELECTIVITY
MOLECULAR AND CELLULAR DETERMINANTS OF DRUG SELECTIVITY
The ideal drug would interact only with a molecular target that causes the desired therapeutic effect but not with molecular targets that cause unwanted adverse effects. Although no such drug has yet been discovered (i.e., all drugs currently in clinical use have the potential to cause adverse effects as well as therapeutic effects; see Chapter 6, Drug Toxicity), pharmacologists can take advantage of several determinants of drug selectivity in an attempt to reach this goal. Selectivity of drug action can be conferred by at least two classes of mechanisms, including (1) the cell-type specificity of receptor subtypes and (2) the cell-type specificity of receptor–effector coupling.
Although many potential receptors for drugs are widely distributed among diverse cell types, some receptors are more limited in their distribution. Systemic administration of drugs that interact with such localized receptors can result in a highly selective therapeutic effect. For example, drugs that target ubiquitous processes such as DNA synthesis are likely to cause significant toxic side effects; this is the case with many currently available chemotherapeutics for the treatment of cancer. Other drugs that target cell-type restricted processes such as acid generation in the stomach may have fewer adverse effects. Imatinib, for example, is an extremely selective drug because the BCR-Abl protein is not expressed in normal (noncancerous) cells. In general, the more restricted the cell-type distribution of the receptor targeted by a particular drug, the more selective the drug is likely to be.
Similarly, even though many different cell types may express the same molecular target for a drug, the effect of that drug may differ in the various cell types because of differential receptor–effector coupling mechanisms or differential requirements for the drug target in the various cell types. For example, although voltage-gated calcium channels are ubiquitously expressed in the heart, cardiac pacemaker cells are relatively more sensitive to the effects of calcium channel blocking agents than are cardiac ventricular muscle cells. This differential effect is attributable to the fact that action potential propagation depends mainly on the action of calcium channels in cardiac pacemaker cells, whereas sodium channels are more important than calcium channels in the action potentials of ventricular muscle cells. In general, the more the receptor–effector coupling mechanisms differ among the various cell types that express a particular molecular target for a drug, the more selective the drug is likely to be.

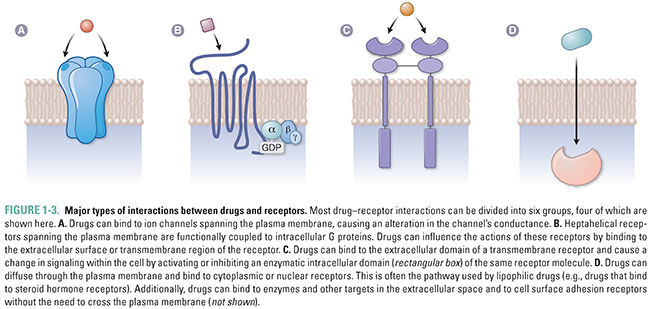
Given the great diversity of drug molecules, it might seem likely that the interactions between drugs and their molecular targets would be equally diverse. This is only partly true. In fact, most of the currently understood drug–receptor interactions can be classified into six major groups. These groups comprise the interactions between drugs and (1) transmembrane ion channels; (2) transmembrane receptors coupled to intracellular G proteins; (3) transmembrane receptors with linked enzymatic domains; (4) intracellular receptors, including enzymes, signal transduction molecules, transcription factors, structural proteins, and nucleic acids; (5) extracellular targets; and (6) cell surface adhesion receptors (Fig. 1-3). Table 1-2 provides a summary of each major interaction type.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


