INTRODUCTION
Our tissues are exposed on a daily basis to xenobiotics—foreign substances that are not naturally found in the body. Most drugs are xenobiotics that are used to modulate bodily functions for therapeutic ends. Drugs and other environmental chemicals that enter the body are modified by a vast array of enzymes. The biochemical transformations performed by these enzymes can alter the compound to render it beneficial, harmful, or simply ineffective. The processes by which biochemical reactions alter drugs within the body are collectively called drug metabolism or drug biotransformation.
The previous chapter introduced the importance of renal clearance in the pharmacokinetics of drugs. Although the biochemical reactions that alter drugs to renally excretable forms are an essential part of drug metabolism, drug metabolism encompasses more than this one function. Drug biotransformation can alter drugs in four important ways:
 An active drug may be converted to an inactive drug.
An active drug may be converted to an inactive drug.
 An active drug may be converted to an active or toxic metabolite.
An active drug may be converted to an active or toxic metabolite.
 An inactive prodrug may be converted to an active drug.
An inactive prodrug may be converted to an active drug.
 An unexcretable drug may be converted to an excretable metabolite (e.g., to enhance renal or biliary clearance).
An unexcretable drug may be converted to an excretable metabolite (e.g., to enhance renal or biliary clearance).
This chapter presents the major processes of drug metabolism. Following the case is an overview of the sites of drug metabolism, focusing principally on the liver. The two major types of biotransformation are then discussed; these are often termed phase I and phase II reactions, although the terminology is imprecise and it incorrectly implies a temporal order of the reactions. (In addition, phase III is sometimes used to describe the process of drug transport.) In this chapter, we use oxidation/reduction and conjugation/hydrolysis to describe these processes more accurately. The chapter concludes with a discussion of the factors that can lead to differences in drug metabolism among individuals.
 Ms. B is a 32-year-old Caucasian woman who complains of sore throat and difficulty swallowing for the past 5 days. Physical examination reveals creamy white lesions on the tongue that are identified as oral thrush, a fungal infection. Her history includes sexual activity with multiple partners, inconsistent use of condoms, and continuous use of oral contraceptives for the past 14 years. The presentation suggests a diagnosis of HIV-1 infection, which is confirmed by polymerase chain reaction (PCR) analysis. Ms. B has a low CD4 T-cell count and is immediately started on a standard anti-HIV regimen that includes the protease inhibitor saquinavir. Her oral thrush resolves with a topical antifungal agent. Despite aggressive therapy, her CD4 cell count continues to decrease, and she presents to her physician several months later with fatigue and a persistent cough. Further investigation leads to a diagnosis of tuberculosis.
Ms. B is a 32-year-old Caucasian woman who complains of sore throat and difficulty swallowing for the past 5 days. Physical examination reveals creamy white lesions on the tongue that are identified as oral thrush, a fungal infection. Her history includes sexual activity with multiple partners, inconsistent use of condoms, and continuous use of oral contraceptives for the past 14 years. The presentation suggests a diagnosis of HIV-1 infection, which is confirmed by polymerase chain reaction (PCR) analysis. Ms. B has a low CD4 T-cell count and is immediately started on a standard anti-HIV regimen that includes the protease inhibitor saquinavir. Her oral thrush resolves with a topical antifungal agent. Despite aggressive therapy, her CD4 cell count continues to decrease, and she presents to her physician several months later with fatigue and a persistent cough. Further investigation leads to a diagnosis of tuberculosis.
Questions
1. One of the first-line drugs in the treatment of tuberculosis is rifampin, which decreases the effectiveness of HIV protease inhibitors. What is the mechanism of this drug–drug interaction?
2. Isoniazid is another drug commonly used in the treatment of tuberculosis. Why does Ms. B’s ethnic background give her physician reason for concern when considering the use of this drug?
3. What dietary interactions should be taken into consideration when prescribing medications to treat Ms. B’s HIV infection?

The liver is the main organ of drug metabolism. This fact figures prominently in the phenomenon known as the first-pass effect. Orally administered drugs are often absorbed in the gastrointestinal (GI) tract and transported directly to the liver via the portal circulation (Fig. 4-1). In this manner, the liver has the opportunity to metabolize drugs before they reach the systemic circulation and, therefore, before they reach their target organs. The first-pass effect must be taken into account when designing dosing regimens because, if hepatic metabolism is extensive, the amount of drug that reaches the target tissue is much less than the amount (dose) that is administered orally (see Chapter 3, Pharmacokinetics). Certain drugs are inactivated so efficiently upon their first pass through the liver that they cannot be administered orally and must be given parenterally. One such drug is the antiarrhythmic lidocaine, which has a bioavailability of only 3% when taken orally (see Chapter 12, Local Anesthetic Pharmacology).
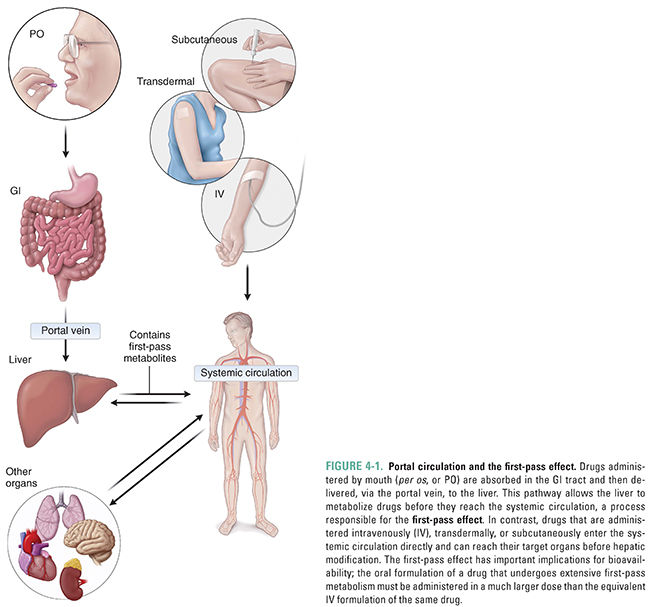
Although the liver is quantitatively the most important organ in metabolizing drugs, every tissue in the body is capable of drug metabolism to some degree. Particularly active sites include the skin, the lungs, the gastrointestinal tract, and the kidneys. The gastrointestinal tract deserves special mention because this organ, like the liver, can contribute to the first-pass effect by metabolizing orally administered drugs before they reach the systemic circulation.

Drugs and other xenobiotics undergo biotransformation before excretion from the body. Many pharmaceuticals are lipophilic, enabling the drug to diffuse across cell membranes, such as those of the intestinal mucosa or of the target tissue. Unfortunately, the same chemical property that enhances bioavailability of drugs may also make renal excretion difficult, because clearance by the kidney requires that these drugs be made more hydrophilic so that they can dissolve in the aqueous urine. Thus, biotransformation reactions often enhance the hydrophilicity of compounds to render them more susceptible to renal excretion.
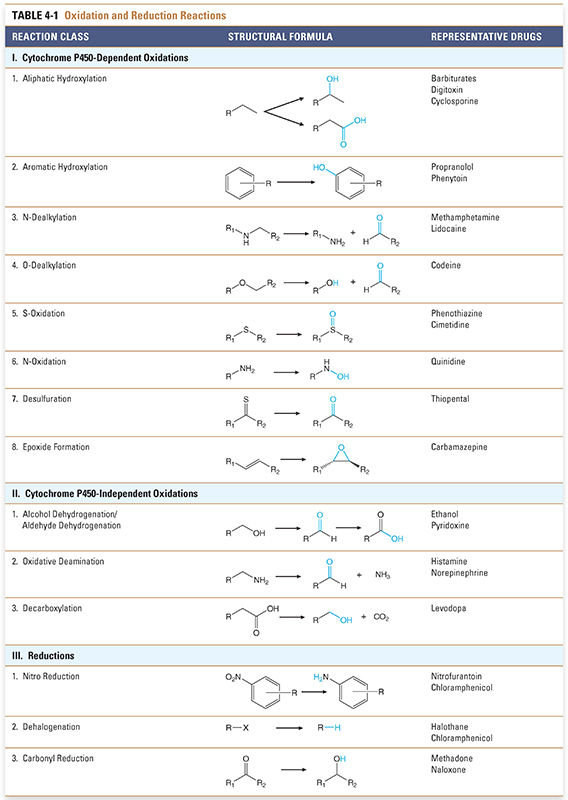
Biotransformation reactions have been classically divided into two main types: oxidation/reduction (phase I) and conjugation/hydrolysis (phase II). Oxidation reactions typically transform the drug into more hydrophilic metabolites by adding or exposing polar functional groups such as hydroxyl (-OH) or amine (-NH2) groups (Table 4-1). Such metabolites are often pharmacologically inactive and, without further modification, may be excreted. Some products of oxidation and reduction reactions, however, require further modifications prior to excretion. Conjugation (phase II) reactions modify compounds through attachment of hydrophilic groups, such as glucuronic acid, to create more polar conjugates (Table 4-2). It is important to note that these conjugation reactions occur independently of oxidation/reduction reactions, and the enzymes involved in oxidation/reduction and conjugation/hydrolysis reactions often compete for substrates.
Oxidation reactions involve membrane-associated enzymes expressed in the endoplasmic reticulum (ER) of hepatocytes and, to a lesser extent, of cells in other tissues. The enzymes that catalyze these phase I reactions are typically oxidases; the majority of these enzymes are heme protein mono-oxygenases of the cytochrome P450 class. Cytochrome P450 enzymes (sometimes abbreviated CYP) are also known as microsomal mixed-function oxidases and are involved in the metabolism of approximately 75% of all drugs used today. (The term P450 refers to the 450-nm absorption peak characteristic of these heme proteins when they bind carbon monoxide.)
The net result of a cytochrome P450-dependent oxidation reaction is:
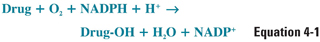
The reaction proceeds when the drug binds to the oxidized (Fe3+) cytochrome P450 to form a complex, which is then reduced in two sequential oxidation/reduction steps as outlined in Figure 4-2A. Nicotinamide adenine dinucleotide phosphate (NADPH) donates the electrons in both of these steps via a flavoprotein reductase. In the first step, the donated electron reduces the cytochrome P450–drug complex. In the second step, the electron reduces molecular oxygen to form an activated oxygen–cytochrome P450–drug complex. Finally, as the complex becomes more active through rearrangement, the reactive oxygen atom is transferred to the drug, resulting in the formation of the oxidized drug product and recycling oxidized cytochrome P450 in the process. The mechanism of these reactions is illustrated in Figure 4-2B.
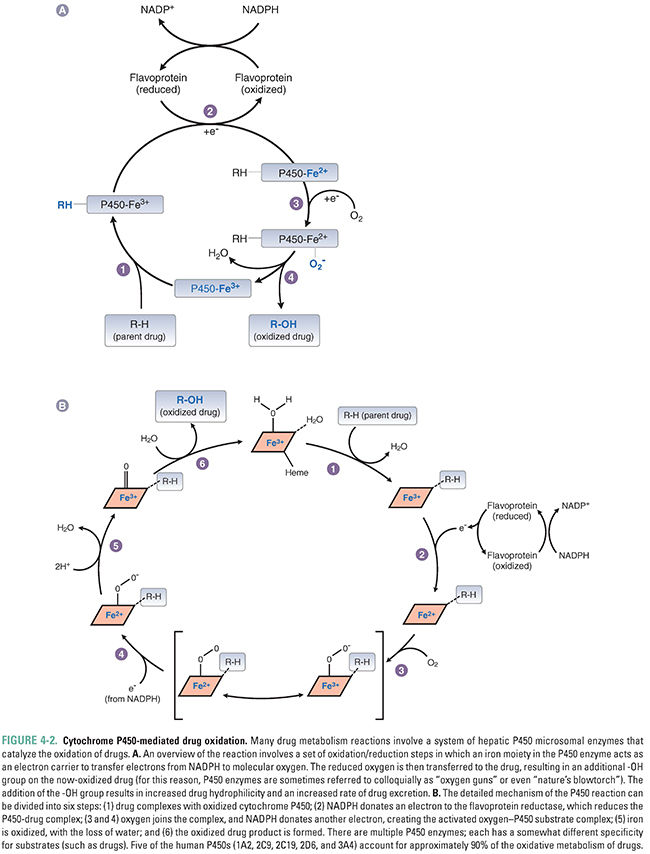
Most liver cytochrome P450 oxidases exhibit broad substrate specificity (Table 4-1). This is due in part to the activated oxygen of the complex, which is a powerful oxidizing agent that can react with a variety of substrates. The names of the cytochrome P450 enzymes are sometimes designated by “P450” followed by the number of the P450 enzyme family, capital letter of the subfamily, and an additional number to identify the specific enzyme (e.g., P450 3A4). Many of the P450 enzymes have partially overlapping specificities that together allow the liver to recognize and metabolize a wide array of xenobiotics.
Together, P450-mediated reactions account for more than 95% of oxidative biotransformations. Other pathways may also oxidize lipophilic molecules. A pertinent example of a non-P450 oxidative pathway is the alcohol dehydrogenase pathway that oxidizes alcohols to their aldehyde derivatives as part of the overall process of excretion. These enzymes are also the basis for the toxicity of methanol. Methanol is oxidized by alcohol dehydrogenase to formaldehyde, which can do considerable damage to some tissues. The optic nerve is particularly sensitive to formaldehyde, and methanol toxicity can cause blindness.
Another important non-P450 enzyme is monoamine oxidase (MAO). This enzyme is responsible for the oxidation of amine-containing endogenous compounds such as catecholamines and tyramine (see Chapter 11, Adrenergic Pharmacology) and some xenobiotics, including drugs.
Conjugation/Hydrolysis Reactions
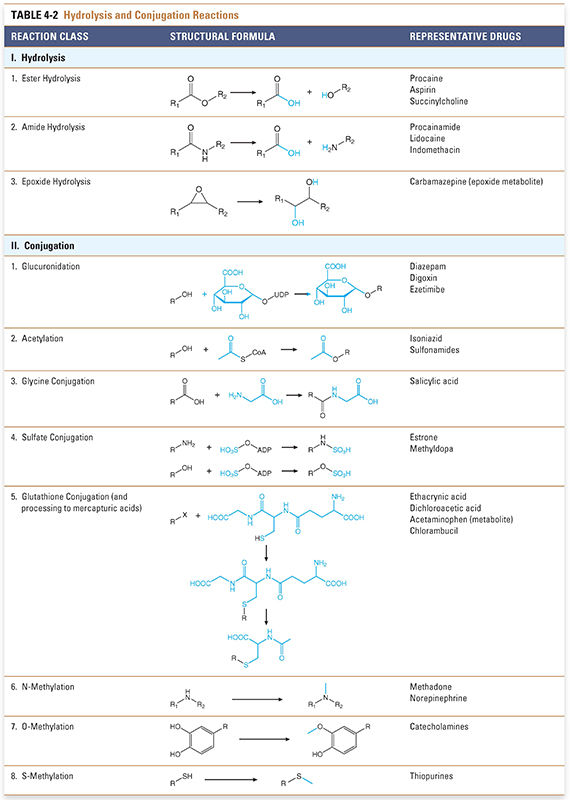
Conjugation and hydrolysis reactions provide a second set of mechanisms for modifying compounds for excretion (Fig. 4-3). Although hydrolysis of ester- and amide-containing drugs is sometimes included among the phase I reactions (in the older terminology), the biochemistry of hydrolysis is more closely related to conjugation than to oxidation/reduction. Substrates for these reactions include both metabolites of oxidation reactions (e.g., epoxides) and compounds that already contain chemical groups appropriate for conjugation, such as hydroxyl (-OH), amine (-NH2), or carboxyl (-COOH) moieties. These substrates are coupled by transfer enzymes to endogenous metabolites (e.g., glucuronic acid and its derivatives, sulfuric acid, acetic acid, amino acids, and the tripeptide glutathione) in reactions that often involve high-energy intermediates (Table 4-2). The conjugation and hydrolysis enzymes are located in both the cytosol and the endoplasmic reticulum of hepatocytes (and other tissues). In most cases, the conjugation process makes the drug more polar. Virtually all of the conjugated products are pharmacologically inactive, with some important exceptions (e.g., morphine glucuronide).
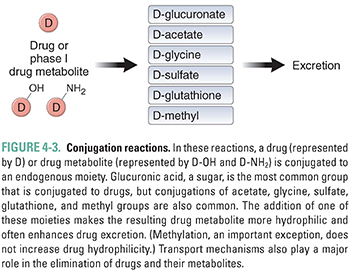
Some conjugation reactions are important clinically in the case of neonates, who have not yet fully developed the capacity to carry out this set of reactions. UDP-glucuronyl transferase (UDPGT) is responsible for conjugating bilirubin in the liver and facilitating its excretion. A relative deficiency of this enzyme at the time of birth puts infants at risk for neonatal jaundice, which results from increased serum levels of unconjugated bilirubin. Neonatal jaundice is a problem because neonates have not only underdeveloped activity of this enzyme but also an undeveloped blood–brain barrier. Unconjugated bilirubin is water-insoluble and very lipophilic; it binds readily to the unprotected neonatal brain and is capable of causing significant damage to the central nervous system. This pathologic condition is known as bilirubin encephalopathy or kernicterus. Neonatal hyperbilirubinemia (unconjugated) can be treated with phototherapy with 450-nm light, which converts circulating bilirubin to an isomer that is more rapidly excreted. Another effective treatment is the administration of small doses of the barbiturate phenobarbital, which powerfully up-regulates the expression of the enzyme UDPGT and thereby reduces serum levels of unconjugated bilirubin. This example illustrates a recurring theme: understanding drug metabolism can help predict both adverse and potentially advantageous drug–drug interactions.
It is important to note that conjugation and hydrolysis reactions do not necessarily constitute the last step of biotransformation. Since the conjugation of these highly polar moieties occurs intracellularly, they often require active transport across cellular membranes to be excreted (active transport of the parent drug can also occur). Moreover, some conjugation products may be subjected to further metabolism.
Although many drugs are sufficiently lipophilic to cross cell membranes passively, it is now appreciated that many drugs need to be transported actively into cells. This fact has significant consequences for oral bioavailability (transport into enterocytes or active excretion into the intestinal lumen), hepatic metabolism (transport into hepatocytes for enzymatic metabolism and for excretion into bile), and renal clearance (transport into proximal tubular cells and excretion into the tubular lumen). Several important molecules mediate these processes. The multidrug resistance protein 1 (MDR1), or P-glycoprotein, which is a member of the ABC family of efflux transporters, actively transports compounds back into the intestinal lumen. This process limits the oral bioavailability of several important drugs, including digoxin and HIV-1 protease inhibitors. The metabolism of drugs from the portal circulation (i.e., the first-pass effect) often requires the transport of compounds into hepatocytes via the organic anion transporting polypeptide (OATP) and the organic cation transporter (OCT) family of proteins. These transporters are particularly relevant for the metabolism of several 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins), which are used in the treatment of hypercholesterolemia. For example, metabolism of the HMG-CoA reductase inhibitor pravastatin is dependent on the transporter OATP1B1, which transports the drug into hepatocytes. Drug uptake into hepatocytes via OATP1B1 is thought to be the rate-limiting step in the clearance of pravastatin from the bloodstream. The uptake of pravastatin on its first pass through the liver also provides a potential advantage by keeping the drug out of the systemic circulation, from which it could be taken up by muscle cells and thereby cause toxic effects such as rhabdomyolysis. The organic anion transporter (OAT) family of transporters is responsible for renal secretion of many clinically important anionic drugs, such as β-lactam antibiotics, nonsteroidal anti-inflammatory drugs (NSAIDs), and antiviral nucleoside analogs.
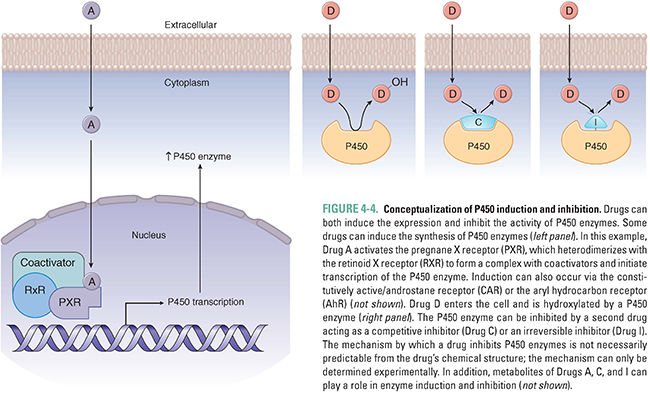
The use of phenobarbital to prevent neonatal jaundice demonstrates that drug metabolism can be influenced by the expression levels of drug-metabolizing enzymes. Although some P450 enzymes are constitutively active, others can be induced or inhibited by various compounds. Induction or inhibition can be incidental (a side effect of a drug) or deliberate (the desired effect of therapy).
The primary mechanism of P450 enzyme induction is an increase in the expression of the enzyme chiefly through increased transcription, although augmented translation and decreased degradation can also have minor roles. The induction of P450 enzymes by a wide array of drugs reflects the biology of xenobiotic receptors that act as the body’s surveillance system to metabolize potentially toxic compounds. Drugs, environmental pollutants, industrial chemicals, and even foodstuffs can enter hepatocytes and bind to several different xenobiotic receptors, such as the pregnane X receptor (PXR), constitutively active/androstane receptor (CAR), and aryl hydrocarbon receptor (AhR) (Fig. 4-4). These molecules are nuclear hormone receptors; when a xenobiotic compound binds to and activates the receptor, the complex is translocated to the nucleus, where it binds to the enhancer regions of various biotransformation enzymes, promoting increased expression of P450 enzymes via transcription. By a similar mechanism, nuclear hormone receptor activation can also increase the expression of drug transporters that aid in clearing the compounds from the body, such as MDR1 and OATP1.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


