15
Diuretic Agents
CASE STUDY
A 65-year-old man has a history of diabetes and chronic kidney disease with baseline creatinine of 2.8 mg/dL. Despite five different antihypertensives, his clinic blood pressure is 176/92 mm Hg and he has 2–3+ edema on exam. He has been taking furosemide 80 mg twice a day for one year now. He has mild dyspnea on exertion. At the clinic visit, hydrochlorothiazide 25 mg daily is added for better blood pressure control and symptoms/signs of fluid overload. Two weeks later, the patient presents to the emergency department with symptoms of weakness, anorexia, and generalized malaise. His blood pressure is now 91/58 mm Hg and he has lost 15 kg in two weeks. His laboratory tests are significant for a serum creatinine of 10.8. What has led to the acute kidney injury? What is the reason for the weight loss? What precautions could have been taken to avoid this hospitalization?
Abnormalities in fluid volume and electrolyte composition are common and important clinical disorders. Drugs that block specific transport functions of the renal tubules are valuable clinical tools in the treatment of these disorders. Although various agents that increase urine volume (diuretics) have been described since antiquity, it was not until 1937 that carbonic anhydrase inhibitors were first described and not until 1957 that a much more useful and powerful diuretic agent (chlorothiazide) became available.
Technically, a “diuretic” is an agent that increases urine volume, whereas a “natriuretic” causes an increase in renal sodium excretion and an “aquaretic” increases excretion of solute-free water. Because natriuretics almost always also increase water excretion, they are usually called diuretics. Osmotic diuretics and antidiuretic hormone antagonists (see Agents that Alter Water Excretion) are aquaretics that are not directly natriuretic.
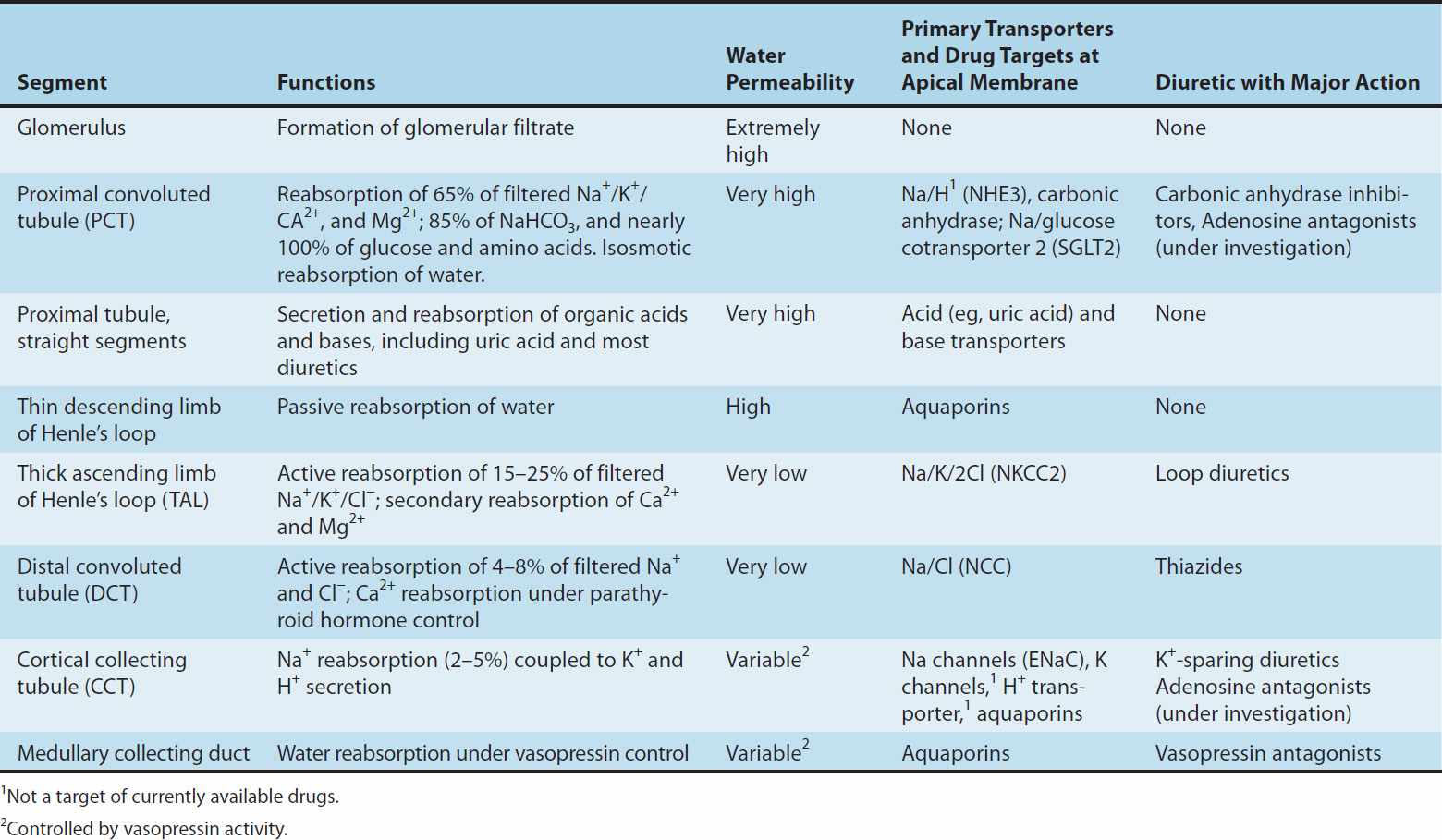
This chapter is divided into three sections. The first section covers major renal tubule transport mechanisms. The nephron is divided structurally and functionally into several segments (Figure 15–1, Table 15–1). Several autacoids, which exert multiple, complex effects on renal physiology (adenosine, prostaglandins, and urodilatin, a renal autacoid closely related to atrial natriuretic peptide), are also discussed. The second section describes the pharmacology of diuretic agents. Many diuretics exert their effects on specific membrane transport proteins in renal tubular epithelial cells. Other diuretics exert osmotic effects that prevent water reabsorption (mannitol), inhibit enzymes (acetazolamide), or interfere with hormone receptors in renal epithelial cells (vaptans, or vasopressin antagonists). The physiology of each nephron segment is closely linked to the basic pharmacology of the drugs acting there, which is discussed in the second section. The third section of the chapter describes the clinical applications of diuretics.
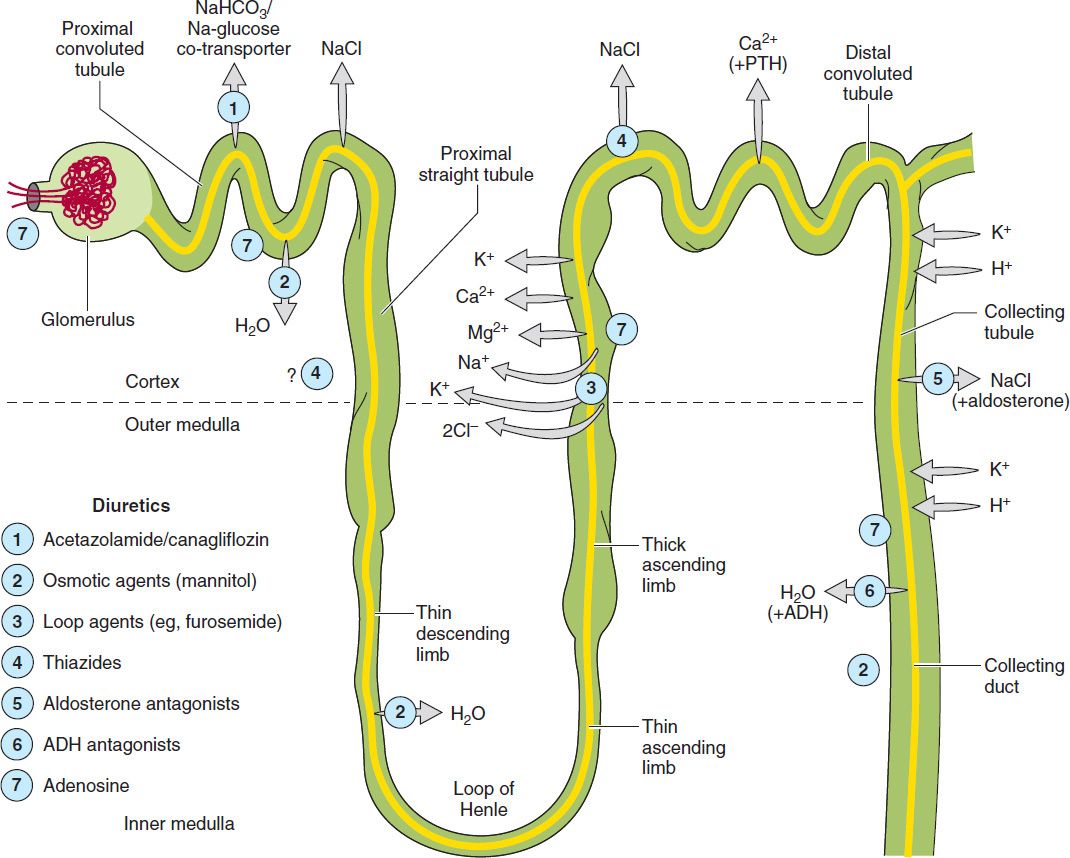
FIGURE 15–1 Tubule transport systems and sites of action of diuretics. ADH, antidiuretic hormone; PTH, parathyroid hormone.
TABLE 15–1 Major segments of the nephron and their functions.
 RENAL TUBULE TRANSPORT MECHANISMS
RENAL TUBULE TRANSPORT MECHANISMS
PROXIMAL TUBULE
Sodium bicarbonate (NaHCO3), sodium chloride (NaCl), glucose, amino acids, and other organic solutes are reabsorbed via specific transport systems in the early proximal tubule (proximal convoluted tubule, PCT). Potassium ions (K+) are reabsorbed via the paracellular pathway. Water is reabsorbed passively, through both a transcellular pathway (mediated by a specific water channel, aquaporin-1 [AQP1]) and a paracellular pathway (likely mediated by claudin-2). Importantly, the water permeability of the PCT is very high, and hence, the osmolality of proximal tubular fluid is maintained at a nearly constant level, and the gradient from the tubule lumen to surrounding interstitium is very small. As tubule fluid is processed along the length of the proximal tubule, the luminal concentrations of most solutes decrease relative to the concentration of inulin, an experimental marker that is filtered but neither secreted nor absorbed by renal tubules. Approximately 66% of filtered sodium ions (Na+), 85% of the NaHCO3, 65% of the K+, 60% of the water, and virtually all of the filtered glucose and amino acids are reabsorbed in the proximal tubule.
Of the various solutes reabsorbed in the proximal tubule, the most relevant to diuretic action are NaHCO3 and NaCl. Until recently, of the currently available diuretics, only one group (carbonic anhydrase inhibitors, which block NaHCO3 reabsorption) has acted predominantly in the PCT. Sodium bicarbonate reabsorption by the PCT is initiated by the action of a Na+/H+ exchanger (NHE3) located in the luminal membrane of the proximal tubule epithelial cell (Figure 15–2). This transport system allows Na+ to enter the cell from the tubular lumen in exchange for a proton (H+) from inside the cell. As in all portions of the nephron, Na+/K+-ATPase in the basolateral membrane pumps the reabsorbed Na+ into the interstitium in order to maintain a low intracellular Na+ concentration. The H+ secreted into the lumen combines with bicarbonate (HCO3−) to form H2CO3 (carbonic acid), which is rapidly dehydrated to CO2 and H2O by carbonic anhydrase. Carbon dioxide produced by dehydration of H2CO3 enters the proximal tubule cell by simple diffusion, where it is then rehydrated back to H2CO3, facilitated by intracellular carbonic anhydrase. After dissociation of H2CO3, the H+ is available for transport by the Na+/H+ exchanger, and the HCO3− is transported out of the cell by a basolateral membrane transporter (Figure 15–2). Bicarbonate reabsorption by the proximal tubule is thus dependent on carbonic anhydrase activity. This enzyme can be inhibited by acetazolamide and other carbonic anhydrase inhibitors.
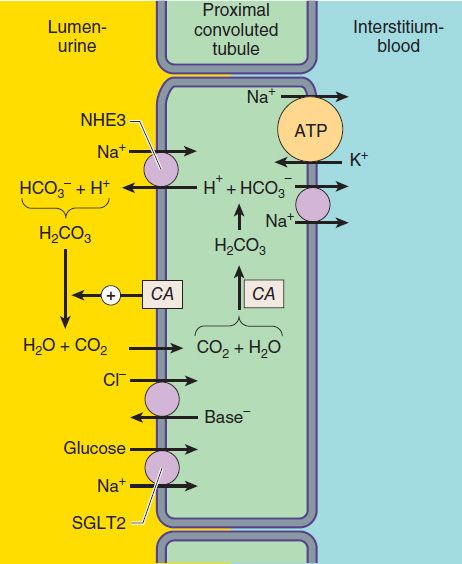
FIGURE 15–2 Apical membrane Na+/H+ exchange (via NHE3) and bicarbonate reabsorption in the proximal convoluted tubule cell. Na+/K+-ATPase is present in the basolateral membrane to maintain intracellular sodium and potassium levels within the normal range. Because of rapid equilibration, concentrations of the solutes are approximately equal in the interstitial fluid and the blood. Carbonic anhydrase (CA) is found in other locations in addition to the brush border of the luminal membrane. SGLT2, Na+/glucose transporter.
More recently, inhibitors of the sodium-glucose cotransporter, isoform 2 (SGLT2; Figure 15–2) have been approved to treat diabetes mellitus. Although not indicated as diuretic agents, these drugs have diuretic properties accompanied by increased sodium and glucose excretion (see below).
Adenosine, which is released as a result of hypoxia and ATP consumption, is a molecule with four different receptors and complex effects on Na+ transport in several segments of the nephron. Although it reduces glomerular filtration rate (GFR) to decrease energy consumption by the kidney, adenosine actually increases proximal reabsorption of Na+ via stimulation of NHE3 activity. A new class of drugs, the adenosine A1-receptor antagonists, have recently been found to significantly blunt both proximal tubule NHE3 activity and collecting duct NaCl reabsorption, and to have potent vasomotor effects in the renal microvasculature (see below, under Autacoids, Pharmacology of Diuretic Agents, and under Heart Failure).
Because HCO3− and organic solutes have been largely removed from the tubular fluid in the late proximal tubule, the residual luminal fluid contains predominantly NaCl. Under these conditions, Na+ reabsorption continues, but the H+ secreted by the Na+/H+ exchanger can no longer bind to HCO3−. Free H+ causes luminal pH to fall, activating a poorly defined Cl−/base exchanger (Figure 15–2). The net effect of parallel Na+/H+ exchange and Cl−/base exchange is NaCl reabsorption. As yet, there are no diuretic agents that are known to act on this conjoint process.
Organic acid secretory systems are located in the middle third of the straight part of the proximal tubule (S2 segment). These systems secrete a variety of organic acids (uric acid, nonsteroidal anti-inflammatory drugs [NSAIDs], diuretics, antibiotics, etc) into the luminal fluid from the blood. These systems thus help deliver diuretics to the luminal side of the tubule, where most of them act. Organic base secretory systems (creatinine, choline, etc) are also present, in the early (S1) and middle (S2) segments of the proximal tubule.
LOOP OF HENLE
At the boundary between the inner and outer stripes of the outer medulla, the proximal tubule empties into the thin descending limb of Henle’s loop. Water is extracted from the descending limb of this loop by osmotic forces found in the hypertonic medullary interstitium. As in the proximal tubule, impermeant luminal solutes such as mannitol oppose this water extraction and thus have aquaretic activity. The thin ascending limb is relatively water-impermeable but is permeable to some solutes.
The thick ascending limb (TAL), which follows the thin limb of Henle’s loop, actively reabsorbs NaCl from the lumen (about 25% of the filtered sodium), but unlike the proximal tubule and the thin descending limb of Henle’s loop, it is nearly impermeable to water. Salt reabsorption in the TAL therefore dilutes the tubular fluid, and it is called a diluting segment. Medullary portions of the TAL contribute to medullary hypertonicity and thereby also play an important role in concentration of urine by the collecting duct.
The NaCl transport system in the luminal membrane of the TAL is a Na+/K+/2Cl−cotransporter (called NKCC2 or NK2CL) (Figure 15–3). This transporter is selectively blocked by diuretic agents known as “loop” diuretics (see later in chapter). Although the Na+/K+/2Cl− transporter is itself electrically neutral (two cations and two anions are cotransported), the action of the transporter contributes to excess K+ accumulation within the cell. Back diffusion of this K+ into the tubular lumen (via the ROMK channel) causes a lumen-positive electrical potential that provides the driving force for reabsorption of cations—including magnesium and calcium—via the paracellular pathway. Thus, inhibition of salt transport in the TAL by loop diuretics, which reduces the lumen-positive potential, causes an increase in urinary excretion of divalent cations in addition to NaCl.
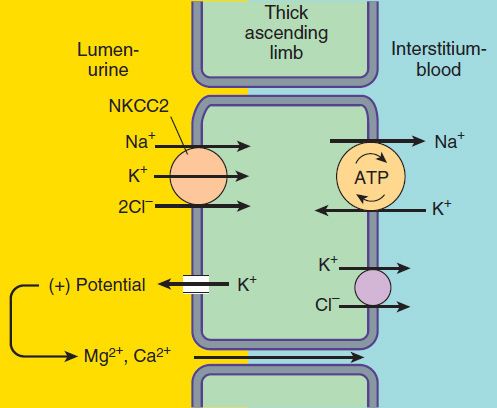
FIGURE 15–3 Ion transport pathways across the luminal and basolateral membranes of the thick ascending limb cell. The lumen positive electrical potential created by K+ back diffusion drives divalent (and monovalent) cation reabsorption via the paracellular pathway. NKCC2 is the primary transporter in the luminal membrane
DISTAL CONVOLUTED TUBULE
Only about 10% of the filtered NaCl is reabsorbed in the distal convoluted tubule (DCT). Like the TAL of Henle’s loop, this segment is relatively impermeable to water, and NaCl reabsorption further dilutes the tubular fluid. The mechanism of NaCl transport in the DCT is an electrically neutral thiazide-sensitive Na+/Cl−cotransporter (NCC ; Figure 15–4).
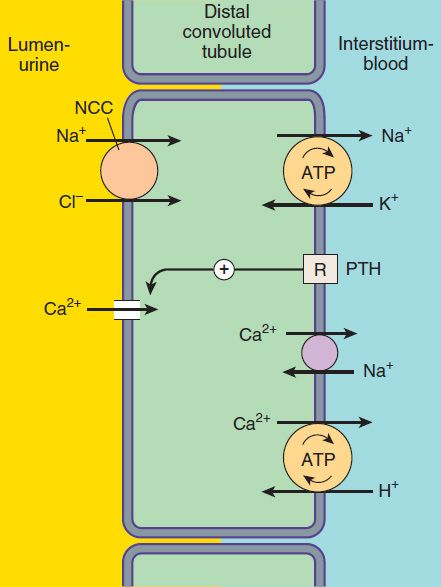
FIGURE 15–4 Ion transport pathways across the luminal and basolateral membranes of the distal convoluted tubule cell. As in all tubular cells, Na+/K+-ATPase is present in the basolateral membrane. NCC is the primary sodium and chloride transporter in the luminal membrane. (R, parathyroid hormone [PTH] receptor.)
Because K+ does not recycle across the apical membrane of the DCT as it does in the TAL, there is no lumen-positive potential in this segment, and Ca2+ and Mg2+ are not driven out of the tubular lumen by electrical forces. Instead, Ca2+ is actively reabsorbed by the DCT epithelial cell via an apical Ca2+ channel and basolateral Na+/Ca2+ exchanger (Figure 15–4). This process is regulated by parathyroid hormone.
COLLECTING TUBULE SYSTEM
The collecting tubule system that connects the DCT to the renal pelvis and the ureter consists of several sequential tubular segments: the connecting tubule, the collecting tubule, and the collecting duct (formed by the connection of two or more collecting tubules). Although these tubule segments may be anatomically distinct, the physiologic gradations are more gradual, and in terms of diuretic activity it is easier to think of this complex as a single segment of the nephron containing several distinct cell types. The collecting tubule system is responsible for only 2–5% of NaCl reabsorption by the kidney. Despite this small contribution, it plays an important role in renal physiology and in diuretic action. As the final site of NaCl reabsorption, the collecting system is responsible for tight regulation of body fluid volume and for determining the final Na+ concentration of the urine. Furthermore, the collecting system is the site at which mineralocorticoids exert a significant influence. Lastly, this is the most important site of K+ secretion by the kidney and the site at which virtually all diuretic-induced changes in K+ balance occur.
The mechanism of NaCl reabsorption in the collecting tubule system is distinct from the mechanisms found in other tubule segments. The principal cells are the major sites of Na+, K+, and water transport (Figures 15–5 and 15–6), and the intercalated cells (α, β) are the primary sites of H+ (a cells) or bicarbonate (β cells) secretion. The α and β intercalated cells are very similar, except that the membrane locations of the H+-ATPase and Cl−/HCO3− exchanger are reversed. Principal cells do not contain apical cotransport systems for Na+ and other ions, unlike cells in other nephron segments. Principal cell membranes exhibit separate ion channels for Na+ and K+. Since these channels exclude anions, transport of Na+ or K+ leads to a net movement of charge across the membrane. Because Na+ entry into the principal cell predominates over K+ secretion into the lumen, a 10–50 mV lumen-negative electrical potential develops. Sodium that enters the principal cell from the tubular fluid is then transported back to the blood via the basolateral Na+/K+-ATPase (Figure 15–5). The 10–50 mV lumen-negative electrical potential drives the transport of Cl− back to the blood via the paracellular pathway and draws K+ out of cells through the apical membrane K+ channel. Thus, there is an important relationship between Na+ delivery to the collecting tubule system and the resulting secretion of K+. Upstream diuretics increase Na+ delivery to this site and enhance K+ secretion. If Na+ is delivered to the collecting system with an anion that cannot be reabsorbed as readily as Cl− (eg, HCO3−), the lumen-negative potential is increased, and K+ secretion is enhanced. This mechanism, combined with enhanced aldosterone secretion due to volume depletion, is the basis for most diuretic-induced K+ wasting. Adenosine antagonists, which act upstream at the proximal tubule, but also at the collecting duct, are perhaps the only diuretics that violate this principle (see below). Reabsorption of Na+ via the epithelial Na channel (ENaC) and its coupled secretion of K+ are regulated by aldosterone. This steroid hormone, through its actions on gene transcription, increases the activity of both apical membrane channels and the basolateral Na+/K+-ATPase. This leads to an increase in the transepithelial electrical potential and a dramatic increase in both Na+ reabsorption and K+ secretion.
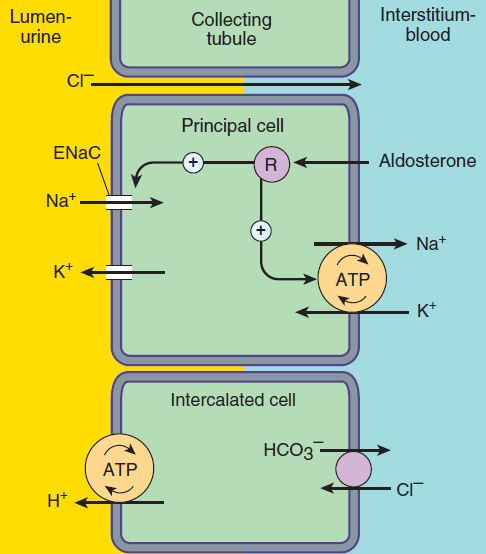
FIGURE 15–5 Ion transport pathways across the luminal and basolateral membranes of collecting tubule and collecting duct cells. Inward diffusion of Na+ via the epithelial sodium channel (ENaC) leaves a lumen-negative potential, which drives reabsorption of Cl− and efflux of K+. (R, aldosterone receptor.)
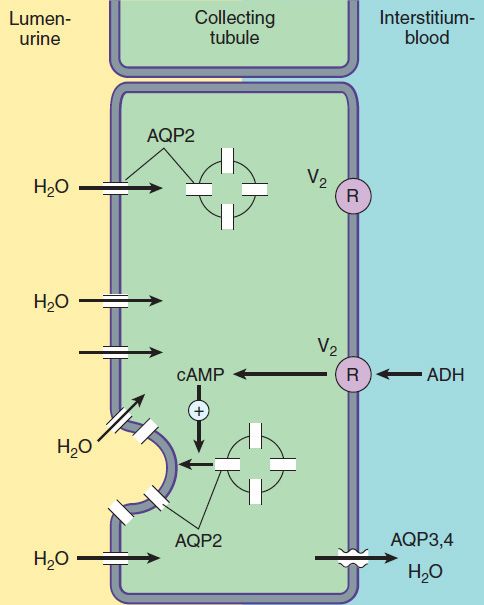
FIGURE 15–6 Water transport across the luminal and basolateral membranes of collecting duct cells. Above, low water permeability exists in the absence of antidiuretic hormone (ADH). Below, in the presence of ADH, aquaporins are inserted into the apical membrane, greatly increasing water permeability. (AQP2, apical aquaporin water channels; AQP3,4, basolateral aquaporin water channels; V2, vasopressin V2 receptor.)
The collecting tubule system is also the site at which the final urine concentration is determined. In addition to their role in control of Na+ absorption and K+ secretion (Figure 15–5), principal cells also contain a regulated system of water channels (Figure 15–6). Antidiuretic hormone (ADH, also called arginine vasopressin, AVP) controls the permeability of these cells to water by regulating the insertion of pre-formed water channels (aquaporin-2, AQP2) into the apical membrane. Vasopressin receptors in the vasculature and central nervous system (CNS) are V1 receptors, and those in the kidney are V2 receptors. V2 receptors act via a Gs protein-coupled, cAMP-mediated process. In the absence of ADH, the collecting tubule (and duct) is impermeable to water, and dilute urine is produced. ADH markedly increases water permeability, and this leads to the formation of a more concentrated urine. ADH also stimulates the insertion of urea transporter UT1 (UT-A, UTA-1) molecules into the apical membranes of collecting duct cells in the medulla.
Urea concentration in the medulla plays an important role maintaining the high osmolarity of the medulla and in the concentration of urine. ADH secretion is regulated by serum osmolality and by volume status. A new class of drugs, the vaptans (see under Agents that Alter Water Excretion), are ADH antagonists.
RENAL AUTACOIDS
A number of locally produced compounds exhibit physiologic effects within the kidney and are therefore referred to as autacoids, or paracrine factors. Several of these autacoids (adenosine, the prostaglandins, and urodilatin) appear to have important effects on the pharmacology of diuretics. Since these effects are complex, they will be treated independently of the individual tubule segments discussed above.
ADENOSINE
Adenosine is an unphosphorylated ribonucleoside whose actions in the kidney have been intensively studied. As in all tissues, renal adenosine concentrations rise in response to hypoxia and ATP consumption. In most tissues, hypoxia results in compensatory vasodilation and, if cardiac output is sufficient, increased blood flow. The kidney has different requirements because increased blood flow leads to an increase in GFR and greater solute delivery to the tubules. This increased delivery would increase tubule work and ATP consumption. In contrast, in the hypoxic kidney, adenosine actually decreases blood flow and GFR. Because the medulla is always more hypoxic than the cortex, adenosine increases Na+ reabsorption from the reduced flow in the cortex, so that delivery to medullary segments will be even further reduced.
There are four distinct adenosine receptors (A1, A2a, A2b, and A3), all of which have been found in the kidney. However, probably only one of these (A1) is of importance with regard to the pharmacology of diuretics. The adenosine A1 receptor is found on the pre-glomerular afferent arteriole, as well as the PCT and most other tubule segments. Adenosine is known to affect ion transport in the PCT, the medullary TAL, and collecting tubules. In addition, adenosine (via A1 receptors on the afferent arteriole) reduces blood flow to the glomerulus (and GFR) and is also the key signaling molecule in the process of tubuloglomerular feedback (see below, under Heart Failure).
In addition to its effects on GFR, adenosine significantly alters Na+ transport in several segments. In the proximal tubule, adenosine has a biphasic effect on NHE3 activity: enhancement at low concentrations and inhibition at very high concentrations. However, adenosine receptor antagonists have generally been found to block the enhancement of NHE3 activity and thus exhibit diuretic activity (see below). It is particularly interesting that unlike other diuretics that act upstream of the collecting tubules, adenosine antagonists do not cause wasting of K+. This important finding suggests that in addition to their effects on NHE3, adenosine antagonists must also blunt K+ secretion in the cortical collecting tubule (CCT). Adenosine A1 receptors have been found in the collecting tubule, but the precise mechanism by which adenosine blunts K+ secretion is not well understood.
PROSTAGLANDINS
Prostaglandins contribute importantly to renal physiology and to the function of many other organs (see Chapter 18). Five prostaglandin subtypes (PGE2, PGI2, PGD2, PGF2α, and thromboxane [TXA2]) are synthesized in the kidney and have receptors in this organ. The role of some of these receptors in renal physiology is not yet completely understood. However, PGE2 (acting on EP1, EP3, and possibly EP2) has been shown to play a role in the activity of certain diuretics. Among its many actions, PGE2 blunts Na+ reabsorption in the TAL of Henle’s loop and ADH-mediated water transport in collecting tubules. These actions of PGE2 contribute significantly to the diuretic efficacy of loop diuretics. Blockade of prostaglandin synthesis with NSAIDs can therefore interfere with loop diuretic activity.
PEPTIDES
There is growing interest in the natriuretic peptides (ANP, BNP, and CNP, see Chapter 17), which induce natriuresis through several different mechanisms. ANP and BNP are synthesized in the heart, while CNP comes primarily from the CNS. Some of these peptides exert both vascular effects (see Chapter 17) and sodium transport effects in the kidney, which participate in causing natriuresis. A fourth natriuretic peptide, urodilatin, is structurally very similar to ANP but is synthesized and functions only in the kidney. Urodilatin is made in distal tubule epithelial cells and blunts Na+ reabsorption through effects on Na+ uptake channels and Na+/K+-ATPase at the downstream collecting tubule system. In addition, through effects on vascular smooth muscle, it reduces glomerular afferent and increases glomerular efferent vasomotor tone. These effects cause an increase in GFR, which adds to the natriuretic activity. Ularitide is a recombinant peptide that mimics the activity of urodilatin. It is currently under intense investigation and may become available for clinical use in the near future.
The cardiac peptides ANP and BNP have pronounced systemic vascular effects. The receptors ANPA and ANPB, also known as NPRA and NPRB, are transmembrane molecules with guanylyl cyclase catalytic activity at the cytoplasmic domains. Of interest, both peptides increase GFR through effects on glomerular arteriolar vasomotor tone and also exhibit diuretic activity. CNP has very little diuretic activity. Three agents in this group are in clinical use or under investigation: nesiritide (BNP), carperitide (ANP, available only in Japan), and ularitide (urodilatin, under investigation). Intravenous ularitide has been studied extensively for use in acute heart failure. It can dramatically improve cardiovascular parameters and promote diuresis without reducing creatinine clearance. There is also evidence that nesiritide (simulating BNP) may enhance the activity of other diuretics while helping to maintain stable renal function. However, the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) study did not show an improvement in outcomes with nesiritide compared with regular care in patients with heart failure.
 BASIC PHARMACOLOGY OF DIURETIC AGENTS
BASIC PHARMACOLOGY OF DIURETIC AGENTS
CARBONIC ANHYDRASE INHIBITORS
Carbonic anhydrase is present in many nephron sites, but the predominant location of this enzyme is the epithelial cells of the PCT (Figure 15–2), where it catalyzes the dehydration of H2CO3 to CO2 at the luminal membrane and rehydration of CO2 to H2CO3 in the cytoplasm as previously described. By blocking carbonic anhydrase, inhibitors blunt NaHCO3 reabsorption and cause diuresis.
Carbonic anhydrase inhibitors were the forerunners of modern diuretics. They were discovered in 1937 when it was found that bacteriostatic sulfonamides caused an alkaline diuresis and hyperchloremic metabolic acidosis. With the development of newer agents, carbonic anhydrase inhibitors are now rarely used as diuretics, but they still have several specific applications that are discussed below. The prototypical carbonic anhydrase inhibitor is acetazolamide.
Pharmacokinetics
The carbonic anhydrase inhibitors are well absorbed after oral administration. An increase in urine pH from the HCO3− diuresis is apparent within 30 minutes, is maximal at 2 hours, and persists for 12 hours after a single dose. Excretion of the drug is by secretion in the proximal tubule S2 segment. Therefore, dosing must be reduced in renal insufficiency.
Pharmacodynamics
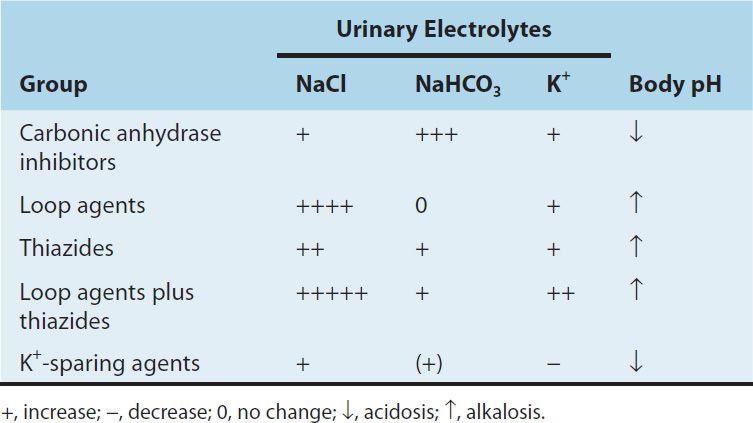
Inhibition of carbonic anhydrase activity profoundly depresses HCO3− reabsorption in the PCT. At its maximal safe dosage, 85% of the HCO3− reabsorptive capacity of the superficial PCT is inhibited. Some HCO3− can still be absorbed at other nephron sites by carbonic anhydrase–independent mechanisms, so the overall effect of maximal acetazolamide dosage is only about 45% inhibition of whole kidney HCO3− reabsorption. Nevertheless, carbonic anhydrase inhibition causes significant HCO3− losses and hyperchloremic metabolic acidosis (Table 15–2). Because of reduced HCO3− in the glomerular filtrate and the fact that HCO3− depletion leads to enhanced NaCl reabsorption by the remainder of the nephron, the diuretic efficacy of acetazolamide decreases significantly with use over several days.
TABLE 15–2 Changes in urinary electrolyte patterns and body pH in response to diuretic drugs.
At present, the major clinical applications of acetazolamide involve carbonic anhydrase–dependent HCO3− and fluid transport at sites other than the kidney. The ciliary body of the eye secretes HCO3− from the blood into the aqueous humor. Likewise, formation of cerebrospinal fluid (CSF) by the choroid plexus involves HCO3− secretion. Although these processes remove HCO3− from the blood (the direction opposite of that in the proximal tubule), they are similarly inhibited by carbonic anhydrase inhibitors.
Clinical Indications & Dosage (Table 15–3)
TABLE 15–3 Carbonic anhydrase inhibitors used orally in the treatment of glaucoma.
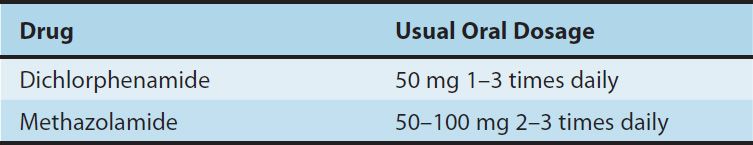
A. Glaucoma
The reduction of aqueous humor formation by carbonic anhydrase inhibitors decreases the intraocular pressure. This effect is valuable in the management of glaucoma in some patients, making it the most common indication for use of carbonic anhydrase inhibitors (see Table 10–3). Topically active agents, which reduce intraocular pressure without producing renal or systemic effects, are available (dorzolamide, brinzolamide).
B. Urinary Alkalinization
Uric acid and cystine are relatively insoluble and may form stones in acidic urine. Therefore, in cystinuria, a disorder of cystine reabsorption, solubility of cystine can be enhanced by increasing urinary pH to 7-7.5 with carbonic anhydrase inhibitors. In the case of uric acid, pH needs to be raised only to 6-6.5. In the absence of HCO3− administration, these effects of acetazolamide last only 2–3 days, so prolonged therapy requires oral HCO3−. Excessive urinary alkalinization can lead to stone formation from calcium salts (see below), so urine pH should be followed during treatment with acetazolamide.
C. Metabolic Alkalosis
Metabolic alkalosis is generally treated by correction of abnormalities in total body K+, intravascular volume, or mineralocorticoid levels. However, when the alkalosis is due to excessive use of diuretics in patients with severe heart failure, replacement of intravascular volume may be contraindicated. In these cases, acetazolamide can be useful in correcting the alkalosis as well as producing a small additional diuresis for correction of volume overload. Acetazolamide can also be used to rapidly correct the metabolic alkalosis that may appear following the correction of respiratory acidosis.
D. Acute Mountain Sickness
Weakness, dizziness, insomnia, headache, and nausea can occur in mountain travelers who rapidly ascend above 3000 m. The symptoms are usually mild and last for a few days. In more serious cases, rapidly progressing pulmonary or cerebral edema can be life-threatening. By decreasing CSF formation and by decreasing the pH of the CSF and brain, acetazolamide can increase ventilation and diminish symptoms of mountain sickness. This mild metabolic central and CSF acidosis is also useful in the treatment of sleep apnea.
E. Other Uses
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


