4 Disorders of growth, differentiation and morphogenesis
Growth, differentiation and morphogenesis are the processes by which a single cell, the fertilised ovum, develops into a large, complex, multicellular organism with co-ordinated organ systems containing a variety of cell types, each with individual specialised functions. Growth and differentiation continue throughout adult life, as many cells of the body undergo a constant cycle of death, replacement and growth in response to normal (physiological) or abnormal (pathological) stimuli.
DEFINITIONS
GROWTH
Types of growth in a tissue (Fig. 4.1A) are:
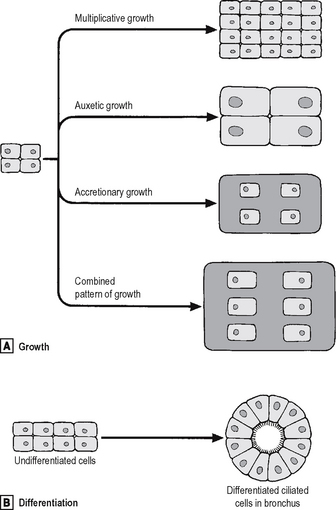
 Types of growth in tissue.
Types of growth in tissue.  Differentiation of undifferentiated cells into ciliated cells in bronchus.
Differentiation of undifferentiated cells into ciliated cells in bronchus.DIFFERENTIATION
Differentiation is the process whereby a cell develops an overt specialised function or morphology which distinguishes it from its parent cell. Thus, differentiation is the process by which genes are expressed selectively and gene products act to produce a cell with a specialised function (Fig. 4.1B). After fertilisation of the human ovum, and up to the eight-cell stage of development, all of the embryonic cells are apparently identical. Thereafter, cells undergo several stages of differentiation in their passage to fully differentiated cells, for example, the ciliated epithelial cells lining the respiratory passages of the nose and trachea. Although the changes at each stage of differentiation may be minor, differentiation can be said to have occurred only if there has been overt change in cell morphology (e.g. development of a skin epithelial cell from an ectodermal cell), or an alteration in the specialised function of a cell (e.g. the synthesis of a hormone).
CELL TURNOVER
In both fetal and adult life, tissue growth depends upon the balance between the increase in cell numbers, due to cell proliferation, and the decrease in cell numbers due to cell death (Fig. 4.2).
REGENERATION
Regeneration enables cells or tissues destroyed by injury or disease to be replaced by functionally identical cells. These replaced ‘daughter’ cells are usually derived from a tissue reservoir of ‘parent’ stem cells (discussed below, page 72). The presence of tissue stem cells, with their ability to proliferate, governs the regenerative potential of a specific cell type. Mammalian cells fall into three classes according to their regenerative ability:
CELL CYCLE
Successive phases of progression of a cell through its cycle of replication are defined with reference to DNA synthesis and cellular division. Unlike the synthesis of most cellular constituents, which occurs throughout the interphase period between cell divisions, DNA synthesis occurs only during a limited period of the interphase: this is the S phase of the cell cycle. A further distinct phase of the cycle is the cell-division stage or M phase (Fig. 4.3) comprising nuclear division (mitosis) and cytoplasmic division (cytokinesis). Following the M phase, the cell enters the first gap (G1) phase and, via the S phase, the second gap (G2) phase before entering the M phase again.
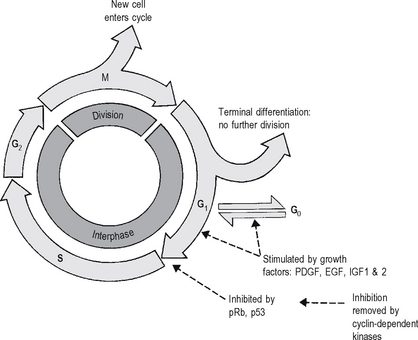
Some cells (e.g. some of the stable cells) may ‘escape’ from the G1 phase of the cell cycle by temporarily entering a G0 ‘resting’ phase: others ‘escape’ permanently to G0 by a process of terminal differentiation, with loss of potential for further division and death at the end of the lifetime of the cell: this occurs in permanent cells, such as neurons.
MOLECULAR EVENTS IN THE CELL CYCLE
At the molecular level, growth is stimulated initially by the receptor-mediated actions of growth factors – e.g. epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and insulin-like growth factors (IGF-1 and IGF-2) – on cells in the quiescent G0 phase of the cell cycle (Fig. 4.3) via intracellular second messengers. Stimuli are transmitted to the nucleus of the cell, where transcription factors are activated, leading to the initiation of DNA synthesis followed by cell division.
The process of cell cycling is modified by the actions of the cyclin family of proteins, which activate (by phosphorylation) a number of proteins involved in DNA replication, mitotic spindle formation and other events in the cell cycle. Thus, for example, the inhibitory (antimitotic) action of the retinoblastoma gene product pRb is itself inhibited by the phosphorylating action of a cyclin-dependent kinase (Fig. 4.3); removal of this growth-inhibiting action of the retinoblastoma gene allows uncontrolled cellular proliferation to proceed, resulting in often rapid growth of this malignant eye neoplasm in children.
DURATION OF THE CELL CYCLE
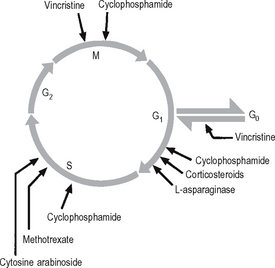
Therapeutic interruption of the cell cycle
Many of the drugs used in the treatment of cancer affect particular stages within the cell cycle (Fig. 4.4). The drugs inhibit the rapid division of cancer cells, although there is often inhibition of other rapidly dividing cells such as the cells of the bone marrow and lymphoid tissues. Thus, anaemia, a bleeding tendency and suppression of immunity may be clinically important side effects of cancer chemotherapy.
CELL DEATH IN GROWTH AND MORPHOGENESIS
APOPTOSIS
Morphogenetic apoptosis
This is involved in alteration of tissue form. Examples include:
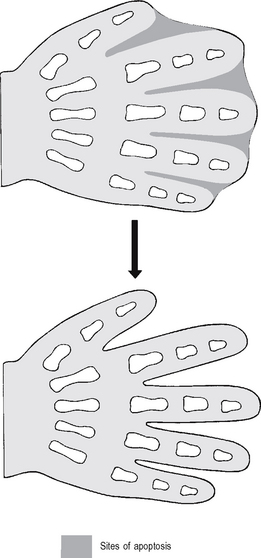
Failure of morphogenetic apoptosis in these four sites is a factor in the development of syndactyly (webbed fingers), cleft palate (see p. 79), spina bifida (see p. 78), and bladder diverticulum (pouch) or fistula (open connection) from the bladder to the umbilical skin.
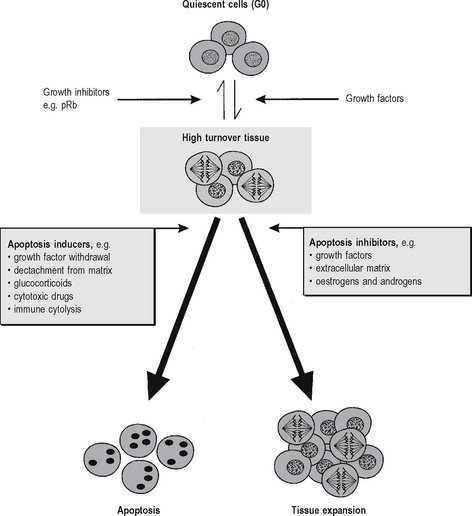
Regulation of apoptosis
When cells within tissues are stimulated to divide by mitogens the tissues enter a high turnover state, in which mitotic activity is accompanied by some degree of coincident apoptosis (Fig. 4.6). The ultimate fate of individual cells within the tissue – whether the cell will survive or undergo apoptosis – depends upon the balance between apoptosis inducers (survival inhibitors) and apoptosis inhibitors (survival factors). Although apoptosis can be induced by diverse signals in a variety of cell types, a few genes appear to regulate a final common pathway. The most important of these are the members of the bcl-2 family (bcl-2 was originally identified at the t(14:18) chromosomal breakpoint in follicular B-cell lymphoma, and it can inhibit many factors which induce apoptosis). The bax protein (also in the bcl-2 family) forms bax-bax dimers which enhance apoptotic stimuli. The ratio of bcl-2 to bax determines the cell’s susceptibility to apoptotic stimuli, and constitutes a ‘molecular switch’ which determines whether a cell will survive (leading to tissue expansion), or undergo apoptosis (leading to tissue contraction).
NORMAL AND ABNORMAL GROWTH IN SINGLE TISSUES
INCREASED GROWTH: HYPERTROPHY AND HYPERPLASIA
The response of an individual cell to increased functional demand is to increase tissue or organ size (Fig. 4.7) by:
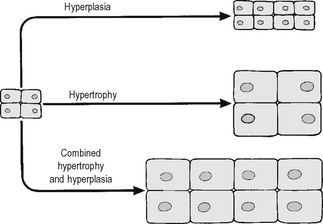
The stimuli for hypertrophy and hyperplasia are very similar, and in many cases identical; indeed, hypertrophy and hyperplasia commonly coexist. In permanent cells (see pp. 13, 53) hypertrophy is the only adaptive option available under stimulatory conditions. In some circumstances, however, permanent cells may increase their DNA content (ploidy) in hypertrophy, although the cells arrest in the G2 phase of the cell cycle without undergoing mitosis; such a circumstance is present in severely hypertrophied hearts, where a large proportion of cells may be polyploid.
An important component of hyperplasia, which is often overlooked, is a decrease in cell loss by apoptosis; the mechanisms of control of this decreased apoptosis are unclear, although they are related to the factors causing increased cell production (Fig. 4.6).
Physiological hypertrophy and hyperplasia
Examples of physiologically increased growth of tissues include:
REPAIR AND REGENERATION
Myofibroblasts
Thus vascular endothelial cell and myofibroblast hyperplasia are important components of repair and regeneration at various sites in the body.
Skin
The healing of a skin wound is a complex process involving the removal of necrotic debris from the wound and repair of the defect by hyperplasia of capillaries, myofibroblasts and epithelial cells. Fig. 4.8 illustrates some of these events, most of which are mediated by growth factors.
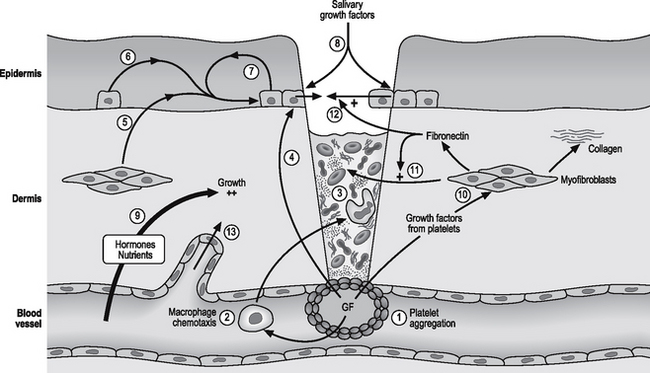
In the epidermis, PDGF acts synergistically with epidermal growth factor (EGF) and the somatomedins (IGF-1 and IGF-2) to promote the progression of basal epithelial cells through the cycle of cell proliferation (p. 53). PDGF acts as a ‘competence factor’ to move cells from their ‘resting’ phase in G0 to G1. EGF and IGFs then act sequentially in cell progression from the G1 phase to that of DNA synthesis. Thereafter, the cell is independent of growth factors. In the epidermis, EGF is derived from epidermal cells (autocrine and paracrine mechanisms), and is also present in high concentrations in saliva when the wound is licked. IGF-1 and IGF-2 originate from the circulation (endocrine mechanisms) and from the proliferating cell and adjacent epidermal and dermal cells (autocrine and paracrine mechanisms).
Ulcers and erosions
Once the ‘ulcerating factor or factors’ are removed, however, the residual ‘survival and healing factors’, or healing capacity of the tissue predominates, and cell proliferation exceeds cell loss, producing net tissue growth to fill the ulcer cavity. In deep ulcers (Fig. 4.9), angiogenic growth factors (produced by macrophages in the necrotic ulcer crater) stimulate growth and migration of capillaries into the base of the ulcer (producing vascular ‘granulation tissue’, seen as finely granular red tissue in the ulcer base). Myofibroblasts also migrate into the ulcer crater, where they proliferate and secrete collagen and matrix proteins, filling the ulcer crater. Once this has happened, the epithelial cells at the edge of the ulcer migrate over the new scar tissue: eventually the ulcer crater is filled, and the epithelium totally covers the former ulcer. Eventually, subepithelial scar tissue contracts (caused by myofibroblast contraction), and myofibroblasts differentiate into mature fibroblasts.
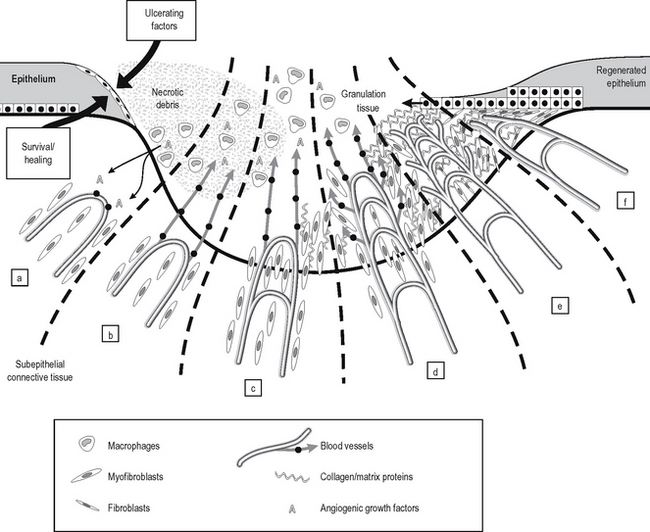
If ‘ulcerating factors’ persist, or if there are recurrent cycles of ulceration – healing – ulceration, an ulcer may become ‘chronic’, with a large deep crater and very extensive scar formation, perhaps leading to marked deformity of the tissue (for example, an ‘hour glass’ deformity with possible stenosis in a stomach with a large chronic gastric ulcer).
Peritoneum
The process of healing and repair of a peritoneal defect is very different to that of an ulcerated epithelial surface, as the mesothelial surface cells do not grow over the defect from its edges. If even large peritoneal defects are left open (not sutured), macrophages migrate into the area to remove necrotic debris (Fig. 4.10). This is followed by a proliferation and migration of peritoneal perivascular connective tissue cells (which resemble primitive mesenchymal cells) into the defect, which eventually fills with these cells. The connective tissue cells on the ‘new’ surface then undergo metaplasia into mesothelial cells. As a result, peritoneal defects heal very rapidly, large defects heal as rapidly as small ones, and peritoneal healing occurs without formation of adhesions.
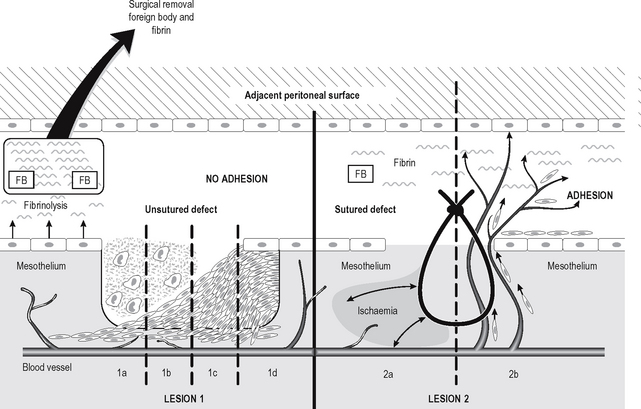
If, however, peritoneal defects are sutured, the suture compresses or tensions the mesothelium and underlying connective tissue, which tends to become relatively ischaemic as a result (Fig. 4.10). As a result, angiogenesis (new blood vessel formation) is stimulated, and capillaries (and later fibroblasts) migrate into the area. If fibrin and/or foreign material such as starch (used to lubricate the inside of surgical gloves) are on the peritoneal surface, the capillaries and fibroblasts grow into the area, and are likely to cause adhesions to adjacent peritoneal surfaces, which may ultimately cause intestinal obstruction. In abdominal and pelvic surgery, therefore, peritoneal surfaces which are left unsutured are less likely to cause adhesions, and both removal of fibrin and prevention of contamination by foreign body materials will reduce the chances of adhesion formation.
Peritoneal mesothelial cells have fibrinolytic activity, but damage to these cells at surgery reduces their ability to remove the peritoneal fibrin which promotes development of adhesions. In addition, growth factors such as epidermal growth factor (EGF) and transforming growth factor beta (TGFβ) may directly influence cell growth in peritoneal healing. However, TGFβ (released in large quantities from platelets at sites of haemorrhage) and tumour necrosis factor (TNF) both probably increase plasminogen-activator inhibitor-1 (PAI-1) activity in peritoneal mesothelial cells, blocking fibrinolytic activity (and hence fibrin removal), and thereby promoting adhesion formation. This is an important field in which further research may well influence the clinical management of patients undergoing abdominal surgery.
Bone
Cellular mechanisms involved in the healing of bone fractures are similar to those in healing in other tissues (Fig. 4.11 illustrates the events involved). Haemor-rhage at the fracture site (inside and around the bone) produces a haematoma, in which there are fragments of necrotic bone, bone marrow, and soft tissues. As is the case in other sites, these necrotic tissues are removed by macrophages. Organisation of the haematoma in bone is accomplished by ingrowth of capillaries and fibroblasts (as in other sites in the body), but is modified in bone by ingrowth of osteoblasts; the resulting proliferation of these cells produces an irregular mass of new irregularly woven bone, called ‘callus’. Internal callus forms within the medullary cavity of the bone; external callus forms in relation to the periosteum, where it acts as a splint until it is finally removed by resorption and remodelling. Eventually, woven bone of the callus is remodelled into lamellar bone, with lamellae oriented according to the direction of mechanical stress on the bone.
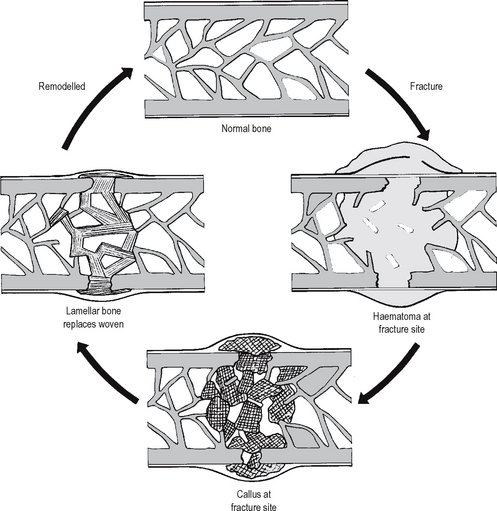
Occasionally bone is lost at the time of fracture (for example, the fractured end of a bone may be removed by the surgeon if heavy contamination has occurred when a compound fracture has penetrated the skin). Under such circumstances the two ends of the bone may be pinned and externally fixed and oriented on an external frame. After initial contact, the bone ends may be gradually separated by increasing traction over several weeks, allowing the bone to be drawn to its correct length whilst the healing process occurs.
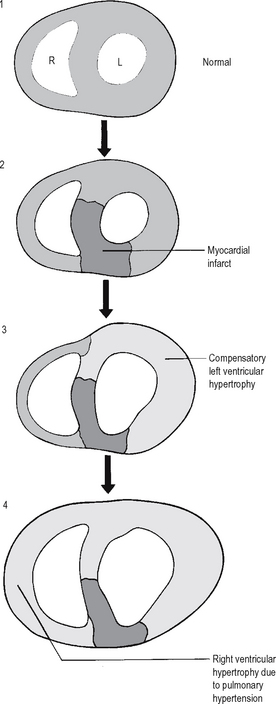
Heart
Myocardial cells are permanent cells and so cannot divide in a regenerative response to tissue injury. In myocardial infarction, a segment of muscle dies and, if the patient survives, it is replaced by hyperplastic myofibroblast scar tissue. As the remainder of the myocardium must work harder for a given cardiac output, it undergoes compensatory hypertrophy (without cell division) (see Fig. 4.12). Occasionally, there may be right ventricular hypertrophy as a result of left ventricular failure and consequent pulmonary hypertension.