1 Cellular injury
All mammalian cells strive to survive against a hostile fluctuating environment by expending energy to maintain a tightly regulated internal and local external environment. If the environmental fluctuations are sufficiently large, they will change the state of the cell, which will then attempt to return to its usual condition. Cellular injury, manifest as a significant disturb-ance of cell function and central to almost all human disease, occurs if the changes in the cell are sufficiently large. In any particular case it may be difficult to tell whether a measured change is due to damage or is due to some meaningful response on the part of the cell.
By cell injury we mean that the cell has been exposed to some influence that has left it living, but functioning at less than optimum level. The end result of this (Fig. 1.1) may be:
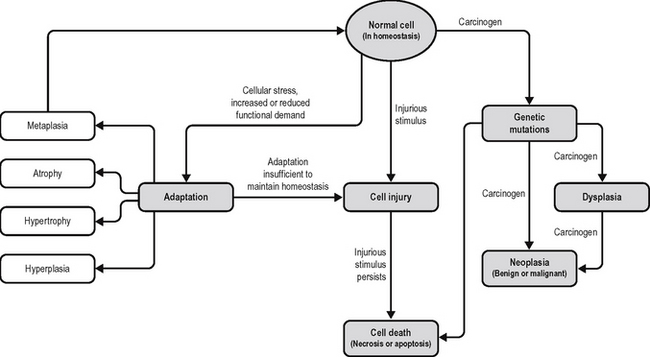
On the whole, (b) is the least likely because cells are capable of significant reparative processes, and if they survive an insult, they generally repair it; if the damage is not lethal but is very severe or persistent and beyond the capacity of the cell to regenerate, the cell may activate mechanisms that result in its own death.
Certain injurious agents (radiation, certain chemicals, viruses, and some bacterial and fungal toxins) directly damage the cell nucleus and deoxyribonucleic acid (DNA), resulting in genetic DNA mutations. Depending on the degree of damage and the portion of the DNA damaged, the damage may be reparable, resulting in a temporary cell cycle arrest but ultimately no phenotypic alteration. Severe irreparable damage triggers apoptotic pathways that culminate in cell death. An intermediate degree of DNA damage results in genetic mutations that do not directly impair cell survival and may confer a survival advantage. Successive mutations will then drive the cell down the multi-step pathway towards neoplasia. The processes involved in oncogenesis are described in Chapter 5.
Cellular injury can be caused by a variety of mechanisms, including:
Cell death may result in replacement by:
MORPHOLOGY OF CELL INJURY
LIGHT MICROSCOPY
Eosinophilic change
Haematoxylin stains acids such as deoxyribonucleic acid (DNA) and ribonucleic-acid (RNA), and eosin stains proteins (proteins are amphoteric but contain many reactive bases). The cytoplasm contains proteins and RNA among other things. Cellular damage often results in a diminution of cytoplasmic RNA, and thus the colour of such cells becomes slightly less purple and more pink (eosinophilic). This is a characteristic of cardiac myocytes in the early stages of ischaemia and may often be the only histologically visible change in postmortem tissue. Eosinophilic change must be distinguished from oncocytosis, which also causes cells to have a profoundly eosinophilic and finely granular cytoplasm due to the accumulation of mitochondria within the cytoplasm. Oncocytic change is seen on occasion as a metaplastic process within the endometrium, but a number of neoplasms including those in the kidney, have oncocytic variants.
ACCUMULATIONS
If a late step in a non-branching metabolic pathway is defective, either genetically or because of some form of trauma, then intermediates earlier in the pathway will accumulate. In some cases where there is branching of the pathway the accumulating materials may be diverted off into alternative processes and the end effect of the insult will be a loss of the usual products occurring after the defective step. Accumulations may be relatively inert, such as lipids occurring in the liver as described above, and their only significance may be as markers of damage. In other cases the accumulated materials may have deleterious effects resulting from direct metabolic influences, e.g. acidosis due to accumulated lactate, or by simple bulk effects such as those seen in various lysosomal storage diseases. Exogenous compounds may be metabolised or stored, but both of these processes may have deleterious consequences. Substances such as carbon tetrachloride are themselves not toxic, but the body has a limited and stereotyped series of responses to external agents and, whilst these responses are on the whole effective at detoxification, in some instances they can result in the production of molecular species more toxic than the original ingested material. In this manner carbon tetrachloride is metabolised in the liver with the production of free radicals which cause severe damage. A similar phenomenon is seen following paracetamol (acetaminophen) overdose. The paracetamol itself is not hepatotoxic, but it is metabolized to n-acetyl p-benzoquinonamine which is potentially hepatotoxic if glutathione levels are depleted. This can be inferred histologically since the liver damage does not occur around the portal vein branches where the carbon tetrachloride or paracetamol enters the liver but only at some distance from this in zones II and III as it becomes metabolised. In the case of ingested asbestos or silica particles, these are taken up into macrophages and cause the disruption of lysosomes, with the release of hydrolytic enzymes. There is consequent minute scarring from this single cell event, but the fibres are then taken up into another macrophage and the process is repeated. Some materials are totally inert, such as carbon, and serve only to show that the individual has a history of exposure to this substance and, more importantly, perhaps to other substances.
Pigments
Exogenous pigments are introduced in tattooing and some have been toxic in various ways. Mercuric chloride (a red pigment) and potassium dichromate (a green pigment) are commonly used in tattooing. Another source for exogenous pigmentation is drugs and organic halogen compounds have often been implicated in abnormal pigmentation problems.
CAUSATIVE AGENTS OF CELL DAMAGE
RADIATION
The problem of variation in energy level of radiation has led to considerable difficulty in establishing suitable measures of dose. The favoured unit currently is the gray (Gy) which is a unit of absorbed dose. One gray is equivalent to 100 rad (the older dose unit of radiation absorbed dose). However, since radiations are often mixed and since tissues have different sensitivities, a mathematically corrected dose called the effective dose equivalent is now used, and the unit of this is the sievert (Sv). The environment contains a number of sources of natural radiation and some degree of contaminant radiation. These include radon liberated from uranium naturally occurring in granite bedrock, and cosmic radiation. The background radiation varies from area to area and with occupations. For example, those frequently engaged in air travel have a higher exposure to cosmic radiation, to which there is approximately a 100 times greater exposure at commercial flight altitudes than at sea level. A pilot flying 600–800 hours per year is exposed to approximately twice the background radiation dose −5 mSv/year – of someone who spends the year at sea level, which is approximately 2.5 mSv/year in the UK. There is considerable debate as to what constitutes a safe level of background radiation or even if there is such a thing as a level of radiation below which no damage will occur. It seems reasonable to assume that no level of radiation can be considered safe no matter how low it is since the safety is only a statistical statement of the likelihood of a mutational event and the probability can never be zero.
DNA damage may have three possible consequences:



