2 Inflammation
Inflammation is the local physiological response to tissue injury. It is not, in itself, a disease, but is usually a manifestation of disease. Inflammation may have beneficial effects, such as the destruction of invading micro-organisms and the walling-off of an abscess cavity, thus preventing spread of infection. Equally, it may produce disease; for example, an abscess in the brain would act as a space-occupying lesion compressing vital surrounding structures, or fibrosis resulting from chronic inflammation may distort the tissues and permanently alter their function.
Inflammation is usually classified according to its time course as:
ACUTE INFLAMMATION
CAUSES OF ACUTE INFLAMMATION
The principal causes of acute inflammation are:
Microbial infections
One of the commonest causes of inflammation is microbial infection. Viruses lead to death of individual cells by intracellular multiplication. Bacteria release specific exotoxins – chemicals synthesised by them which specifically initiate inflammation – or endotoxins, which are associated with their cell walls. Additionally, some organisms cause immunologically-mediated inflammation through hypersensitivity reactions (Chapter 6). Parasite infections and tuberculous inflammation are instances where hypersensitivity is important.
Hypersensitivity reactions
A hypersensitivity reaction occurs when an altered state of immunological responsiveness causes an inappropriate or excessive immune reaction which damages the tissues. The types of reaction are classified in Chapter 6 but all have cellular or chemical mediators similar to those involved in inflammation.
ESSENTIAL MACROSCOPIC APPEARANCES OF ACUTE INFLAMMATION
Swelling (tumor)
Swelling results from oedema – the accumulation of fluid in the extravascular space as part of the fluid exudate – and, to a much lesser extent, from the phys-ical mass of the inflammatory cells migrating into the area (Fig. 2.1).
EARLY STAGES OF ACUTE INFLAMMATION
Changes in vessel calibre
The microcirculation consists of the network of small capillaries lying between arterioles, which have a thick muscular wall, and thin-walled venules. Capillaries have no smooth muscle in their walls to control their calibre, and are so narrow that red blood cells must pass through them in single file. The smooth muscle of arteriolar walls forms precapillary sphincters which regulate blood flow through the capillary bed. Flow through the capillaries is intermittent, and some form preferential channels for flow while others are usually shut down (Fig. 2.2).
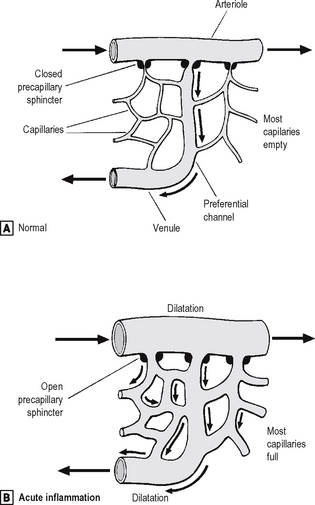
Fig. 2.2 Vascular dilatation in acute inflammation.
 Normally, most of the capillary bed is closed down by precapillary sphincters.
Normally, most of the capillary bed is closed down by precapillary sphincters.  In acute inflammation, the sphincters open, causing blood to flow through all capillaries.
In acute inflammation, the sphincters open, causing blood to flow through all capillaries.Source: Stephenson T J, Inflammation. In: Underwood J C E (ed) General and systemic pathology, 4th edn, Churchill Livingstone, Edinburgh (2004).
Increased vascular permeability
Small blood vessels are lined by a single layer of endothelial cells. In some tissues, these form a complete layer of uniform thickness around the vessel wall, while in other tissues there are areas of endothelial cell thinning, known as fenestrations. The walls of small blood vessels act as a microfilter, allowing the passage of water and solutes but blocking that of large molecules and cells. Oxygen, carbon dioxide and some nutrients transfer across the wall by diffusion, but the main transfer of fluid and solutes is by ultrafiltration, as described by Starling. The high colloid osmotic pressure inside the vessel, due to plasma proteins, favours fluid return to the vascular compartment. Under normal circumstances, high hydrostatic pressure at the arteriolar end of capillaries forces fluid out into the extravascular space, but this fluid returns into the capillaries at their venous end, where hydrostatic pressure is low (Fig. 2.3). In acute inflammation, however, not only is capillary hydrostatic pressure increased, but there is also escape of plasma proteins into the extravascular space, increasing the colloid osmotic pressure there. Consequently, much more fluid leaves the vessels than is returned to them. The net escape of protein-rich fluid is called exudation; hence, the fluid is called the fluid exudate.
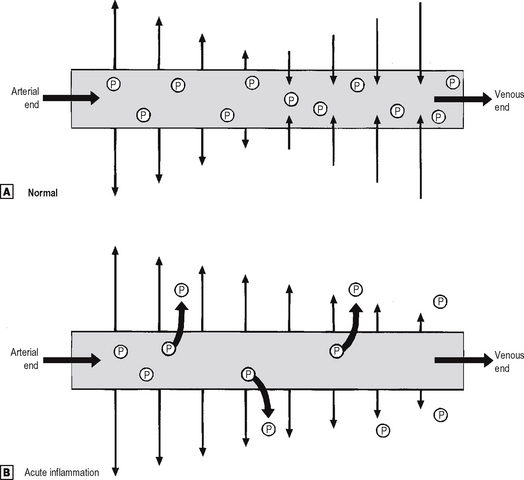
 Normally, fluid leaving and entering the vessel is in equilibrium.
Normally, fluid leaving and entering the vessel is in equilibrium.  In acute inflammation, there is a net loss of fluid together with plasma protein molecules (P) into the extracellular space, resulting in oedema.
In acute inflammation, there is a net loss of fluid together with plasma protein molecules (P) into the extracellular space, resulting in oedema.Formation of the fluid exudate
The increased vascular permeability means that large molecules, such as proteins, can escape from vessels. Hence, the exudate fluid has a high protein content of up to 50 g/l. The proteins present include immunoglobu-lins, which may be important in the destruction of invading micro-organisms, and coagulation factors, including fibrinogen, which result in fibrin deposition on contact with the extravascular tissues. Hence, acute inflamed organ surfaces are commonly covered by fibrin: the fibrinous exudate. There is a considerable turnover of the inflammatory exudate; it is constantly drained away by local lymphatic channels to be replaced by new exudate.
Other causes of increased vascular permeability
In addition to the transient vascular leakage caused by some inflammatory stimuli, certain other stimuli, e.g. heat, cold, ultraviolet light and x-rays, bacterial toxins and corrosive chemicals, cause delayed prolonged leakage. In these circumstances, there is direct injury to endothelial cells in several types of vessels within the damaged area (Table 2.1).
Table 2.1 Causes of increased vascular permeability
| Time course | Mechanisms |
|---|---|
| Immediate transient | Chemical mediators, e.g. histamine, bradykinin, nitric oxide, C5a, leukotriene B4, platelet activating factor |
| Immediate sustained | Severe direct vascular injury, e.g. trauma |
| Delayed prolonged | Endothelial cell injury, e.g. x-rays, bacterial toxins |
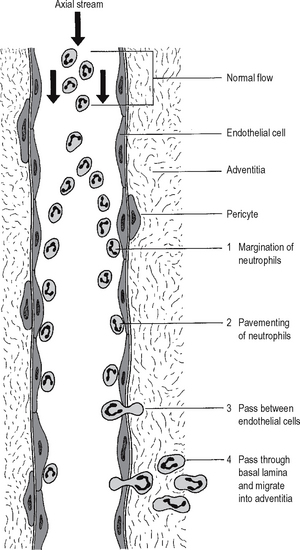
Neutrophil emigration
Chemotaxis of neutrophils
It has been long known from in vitro experiments that neutrophil polymorphs are attracted towards certain chemical substances in solution – a process called chemotaxis. Video microscopy shows apparently purposeful migration of neutrophils along a concentration gradient. Compounds which appear chemotactic for neutrophils in vitro include certain complement components, cytokines and products produced by neutrophils themselves. It is not known whether chemo-taxis is important in vivo. Neutrophils may possibly arrive at sites of injury by random movement, and then be trapped there by immobilising factors (a process analogous to the trapping of macrophages at sites of delayed type hypersensitivity by migration inhibitory factor; Chapter 6).
CHEMICAL MEDIATORS OF ACUTE INFLAMMATION
Chemical mediators released from cells
Leukotrienes
These are also synthesised from arachidonic acid, especially in neutrophils, and appear to have vasoactive properties. SRS-A (slow reacting substance of anaphylaxis), involved in type I hypersensitivity (Chapter 6), is a mixture of leukotrienes.
5-hydroxytryptamine (serotonin)
This is present in high concentration in mast cells and platelets. It is a potent vasoconstrictor.
Plasma factors
Complement system
The complement system is a cascade system of enzym-atic proteins (Chapter 6). It can be activated during the acute inflammatory reaction in various ways:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


