6 Immunology
THE IMMUNE RESPONSE
Immunity is defined as resistance to disease. At its most basic level this consists of a physical barrier (the mucosal surfaces and skin) and the antibacterial actions of certain components of secretions such as lacto-ferrin and enzymes. Much more effective defences are accomplished by two systems which amplify and focus inflammatory responses onto invading foreign substances (or damaged self components). Basic immune responses can be divided into innate (non-adaptive) and specific (adaptive) effector mechanisms (Fig. 6.1).
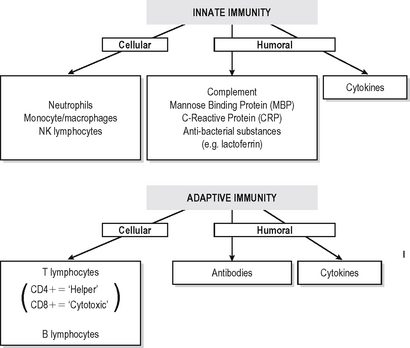
INNATE IMMUNITY
Innate immune defences consist of:
Recently it has been recognised that phagocytic effector cells of the innate immune system such as macrophages, dendritic cells and NK lymphocytes recognize pathogen associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) which are germ-line encoded proteins on pathogens. (Table 6.1). The most important of these PRRs are the toll-like receptors (TLRs), although others exist.
Table 6.1 Pattern recognition of receptors of innate immune system
| PAMPs | PRR’s |
|---|---|
| Gram Negative Organism | |
| LPS Lipopolysacharride | TLR – 4 |
| Gram Positive Organism | |
| Lipoteichoic acid | |
| Lipoarabinomannan | TLR – 2, TLR – 6 |
| RNA | TLR – 7 |
| CPG DNA | TLR – 9 |
| Mannans | Macrophage mannose receptor |
| Lectins | CRP |
SPECIFIC IMMUNE RESPONSES
Specific (adaptive) immune responses are more effective than innate ones and are mediated by lymphocytes and antibodies which amplify and focus non-specific responses and provide additional effector functions. These cells are organised into lymphoid tissues (Fig. 6.2). Humoral immunity often refers to the antibody arm of the specific immune response. Cellular (cell-mediated) immunity refers to lymphocyte-mediated effector responses (T helper (Th) and cytotoxic cells) of the specific immune response. These two arms of the specific immune response are not really separ-able, since antibodies are usually not produced without some cell-mediated response to the same antigen and vice-versa. T and B lymphocytes possess infinitely variable antigen receptors which can clonally expand. Antigen receptors which can be secreted into interstitial fluid and onto mucosal surfaces are called antibodies. Antibodies can activate complement and also enhance opsonisation of antigen to facilitate phagocytosis. Both innate and adaptive mechanisms exponentially amplify the immune response, since clonal expansion of lymphocytes increases the number of cells reactive with an antigen. Cytokines and complement compon-ents recruit other immune effector mechanisms and antibodies activate complement and phagocytes.
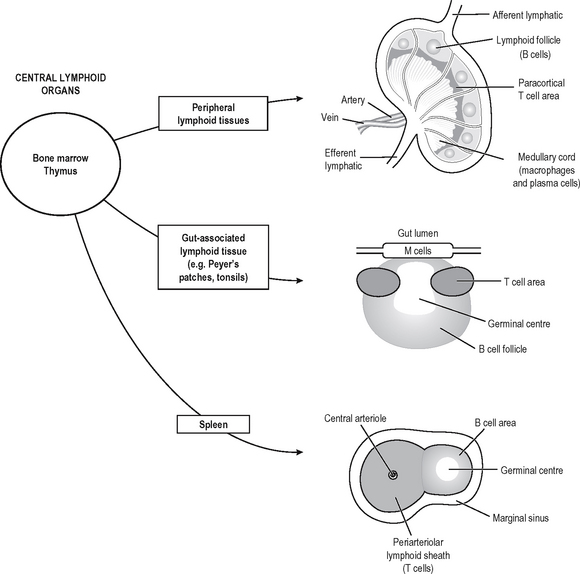
IMMUNE RESPONSE
The components of each type of response in the prevention of individual infections or in the pathogenesis of autoimmune diseases differs (Table 6.2).
Table 6.2 Infections and protective immune responses
| Type of infectious agent | Immune response |
|---|---|
| Extracellular bacteria | Antibodies |
| Pneumococcus | Complement |
| Meningococci | Phagocytes |
| Intracellular bacteria | Antibodies |
| Salmonella | Phagocytes |
| Brucella | CD4 T cells |
| Intracellular Mycobacteria | Activated macrophages |
| Type I cytokines | |
| Viruses | Mucosal protection by IgA antibodies |
| CD8 T cells | |
| NK cells | |
| Parasites | |
| Extracellular | Eosinophils, Macrophages Cytokines IL2, IL4, IL5 IgE |
| Intracellular | CD4 & CD8 T lymphocytes |
COMPLEMENT
The complement system is a soluble enzymic cascade which focuses and amplifies the activity of the specific and innate immune systems as well as having lytic activity against bacteria (Fig. 6.3). It is part of the innate defences since it has no intrinsic antigen specificity.
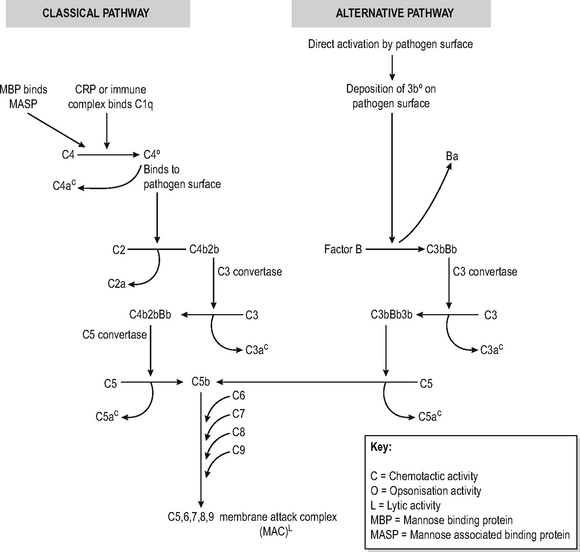
The complement cascade has a final common path-way which leads to the insertion of a multimeric pore-forming structure (membrane associated complex (MAC) consisting of complement components C5-9) into bacterial cell membranes, leading to osmotic lysis. The production of this lytic complex is achieved via two mechanisms called the classical and alternative pathways. Inability to generate the MAC complex leads to particular susceptibility to infections with Neisserial organisms causing recurrent meningitis.
Classical pathway
The classical pathway is triggered by antigen-antibody immune complexes which bind circulating complement factor C1q to the Fc region of the antibody tail, which has undergone conformational changes as a result of antibody binding. The resultant sequential activation of complement proteins results in the formation of a C3 convertase (C4b2b) which cleaves C3, thus forming a C5-convertase (C4b2b3b) which catalyses the production of the C5-9 pore-forming complex. In the process, C2, C3 and C4 are split into fragments, the smaller of which (C2a, C3a, C4a) are chemotactic and the larger of which (C3b, C4b) bind to immune complexes to opsonise or solubilise them, or to a pathogen surface to opsonise it. Thus multiple effects ensuing on other effector mechanisms are caused as a result of complement activation. CRP and MBL can directly activate the classical pathway of complement without the intervention of immune complexes. The lectin pathway is very similar to classical pathway complement, with MBL binding to mannose on pathogens, which is then sequentially bound by MASP to form a C3 convertase. Deficiencies of early complement components are associated with increased risk of developing autoimmune and immune complex disease, possibly because of inability to solubilise immune complexes.
ANTIBODIES
An antibody is a soluble protein immune receptor produced by B lymphocytes, consisting of two identical antigen-binding sites (Fig. 6.4). The antigen specificity of the antibody resides in the antigen-binding variable regions (the fragment antigen-binding, Fab, portion). Antibodies are divided into different isotypes (classes) which have different functional attributes due to the Fc (fragment constant) tails coded by the constant region genes of the heavy chain; thus different constant region genes produce different antibody classes. Antibodies which bind to antigen or cells and activate complement via the Fc region thus recruit, activate, amplify and target non-specific defence mechanisms.
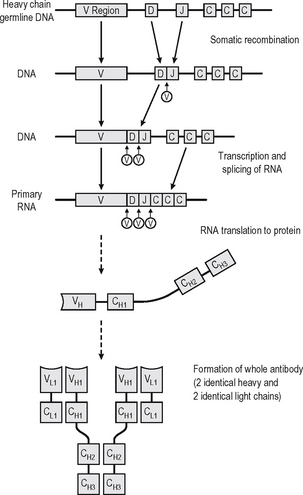
Up to 1010 different antibody specificities may be produced in any individual. This is achieved by joining multiple different copies of genes encoding the variable regions of heavy and light chains of the immunoglobin. Somatic recombination of the gene segments (V, D and J region genes) leads to generation of diversity and broad repertoire of antibody specificities. The antigen-binding variable regions are further (infinitely) diversi-fied by a combination somatic hypermutation (Fig. 6.4) which results from random mutations to the V genes in the hypervariable regions (mutation hotspots) and to the joins between V, D and J genes, enabling antibodies to be produced which can bind to virtually any natural or synthetic antigen encountered. Each cell producing antibody which binds an epitope of an antigen is stimulated to clonally reproduce, and thus further amplification of the immune response occurs with the progeny of each cell producing exactly the same antibody but many different clones expanding.
Isotypes and subclasses
B lymphocytes initially produce IgM upon a primary encounter with antigen; this is very efficient at complement fixation and opsonisation, but IgM circulates as a large pentameric (five antibody molecules) structure with a short half-life (∼five days). Subsequently an individual B cell will undergo a class-switch to IgG, IgA, or IgE production, but class-switching depends on effective T cell help following T cell recognition of an epitope on the same antigen. Memory develops in parallel with the class switch. Both these processes require effective communication between B-cells, Antigen Presenting Cells (APC) and T cells (mediated by CD40– CD40L interaction). IgG diffuses well into extracellular spaces and can neutralise circulating viruses and bacteria (prevent binding by blocking receptors), opsonise via complement or Fc receptors or lyse via complement activation. IgG is divided into four subclasses (IgG1, IgG2, IgG3, IgG4) which have different Fc regions (and thus are coded by different heavy chain constant region gene segments). These classes and subclasses have different half lives and abilities to fix complement, or bind Fc receptors (Table 6.3).
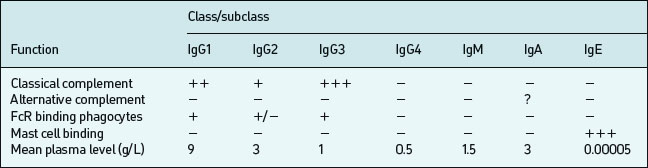
There are several different types of Fc receptors (FcRI or CD64, FcRII or CD32, FcRIII or CD16) which bind some IgG subclasses better than others and are distributed differently on each effector cell type. IgG1 constitutes 60–70% of the circulating IgG in man; IgG2 constitutes 20–40%. IgG3 constitutes 15–20%. IgG4 circulates in trace amounts and its functional significance is unknown, although it may be important in IgE-mediated antiparasite and allergic responses. IgG1 and IgG3 tend to be produced in response to protein antigens; IgG2 in response to polysaccharide antigens (such as those of bacterial capsules).
ANTIGEN-PRESENTING CELLS
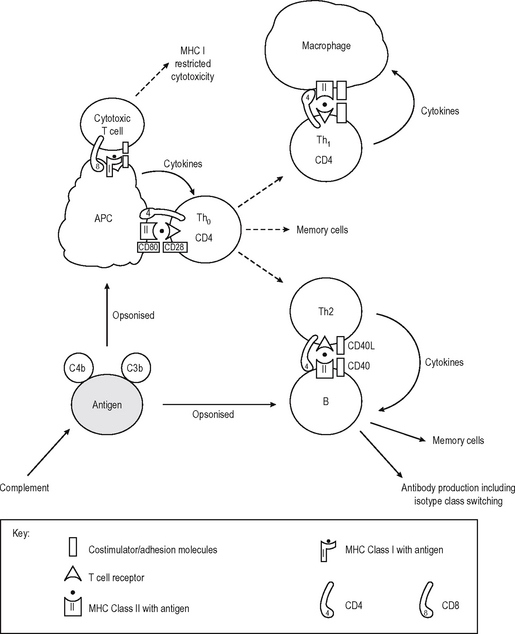
In contrast to antibodies, T cells can not recognize native antigens. They recognise short linear peptides on the surface of APC which digest the whole antigen and present the fragments on the surface in the grooves of major histocompatibility complex (MHC) Class I or II molecules (MHC restriction). The initial interaction of T – lymphocytes with antigen is important in determining whether a specific immune response is promoted or suppressed. The default pathway in unprimed ‘naive’ cells (which have not encountered specific antigen before) is either to become specifically unresponsive to the antigen (anergy) or to die (apoptosis) if the antigen is encountered in an insufficiently stimulating context. Naive T lymphocytes are relatively refractory to stimulation, and require potent signals to activate them to clonally proliferate and/or become effector cells. This usually occurs centrally in the lymph nodes, bone marrow or spleen, but can occur elsewhere. These extra signals are complex and multifactorial but act in addition to the recognition of antigenic peptide in the MHC groove by the T cell receptor (TCR) on the CD4 or the CD8 T cell. This incorporates adhesion molecules which stabilise contact between lymphocyte and APC, and costimulator molecules which provide activation signals to the T cell from the APC (Immunological Synapse – cf. neuro-logical synapse). Important interactions occurring at the immunological synapse are shown in Table 6.4.
Table 6.4 Immunological Synapse – crosstalk between APC and T cell
| TCR | ↔ | Antigen peptide MHC groove |
| CD4 | ↔ | MHC II |
| CD8 | ↔ | MHC I |
| CD 28 (Stimulating) | ↔ | CD 80/CD86 |
| CTLA 4 (Inhibiting) | ↔ | CD80/CD86 |
| CD2 | ↔ | CD58 (LFA3) |
| CD40L | ↔ | CD40 |
APC of several different types provide these second signals while presenting a processed fragment of antigen to a lymphocyte (Fig. 6.5). Primary stimulation of naive T cells requires a potent professional APC (such as the Dendritic cell (DC) or an activated B lymphocyte) with potent stimulatory capacity and ability to acquire and process (digest) antigen by phagocytosis or endocytosis. Secondary restimulation of recently activated or memory T cells is less stringent and can occur on non-professional APC which are not potent enough to stimulate naive cells effectively, e.g. activated endothelium or monocytes and other cells expressing MHC Class II molecules.
T LYMPHOCYTES
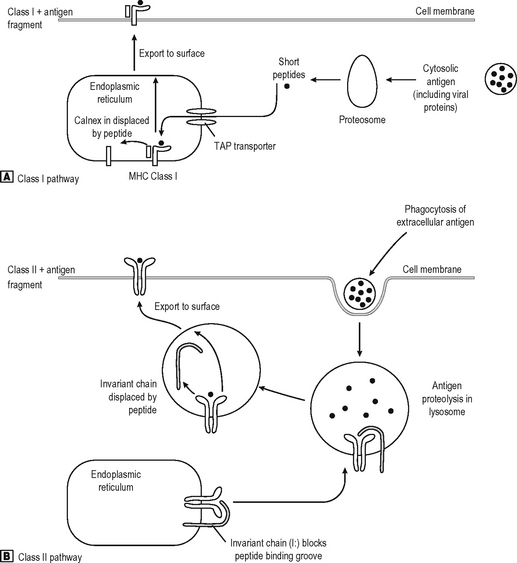
T cells recognise antigen fragments on the surface of APC which express MHC Class I and II molecules on the surface. MHC molecules have an antigen-binding groove on the surface which can bind antigen fragments of 9–11 amino acids (MHC Class I) or 14–20 amino acids (MHC Class II) in length. Thus they act as display platforms on which the TCR can recognise antigen, but because they bind antigen fragments themselves, the MHC molecules also influence the immune responses in any individual since each MHC type will bind some antigens better than others, and occasionally won’t bind some antigens at all. The TCR binds to a part of the lips of the groove as well as the antigen fragment. Thus the TCR is also self-restricted (MHC Restriction), since it binds only to the combination of [self antigen (MHC) + foreign antigen]. A T cell will not operate effectively with non-self APC which bears different MHC molecules. They can, however, co-operate with non-self cells provided they express the same MHC molecules (as they have to do in allogeneic bone marrow transplantation where the BM is donor-type and the recipient is host-type). MHC Class I is bound by CD8, and MHC Class II by CD4 on the T cell surface (Fig. 6.5). Virtually every nucleated cell expresses MHC Class I on the surface, but MHC Class II expression is restricted to certain cell types (e.g. Professional APC, B lymphocytes) especially when the cell is activated. APC express MHC Class II in high density and thus are the major activators of CD4 positive lymphocytes. MHC Class I restricted CD8 positive T cells are also stimulated by APC, but they recognize foreign peptides (e.g. viral, intracellular bacteria) on all nucleated cells by ‘seeing’ viral antigen in the surface groove of self-MHC Class I, and are activated to deliver a lethal attack on the cell. Not surprisingly, viruses have adapted to reduce MHC Class I surface expression (e.g. adenovirus) and can partially evade their attentions (NB NK cells recognize this lack of MHC class I as a sign of an infected cell). Degraded intracellular antigens in the cytosol tend to get access to the MHC Class I groove in the process of MHC assembly in the endoplasmic reticulum, and thus responses to intracellular antigens tend to occur via the MHC Class I pathway (Fig. 6.6). Extracellular antigens from bacteria phagocytosed and digested in the lysosomes of APC tend to gain access to MHC Class II most (readily since the assembly pathway of MHC Class II molecules intersects with the lysosomal pathway). Thus degraded extracellular antigen gains access to ‘empty’ MHC Class II molecules after the invariant chain (which occupies the MHC groove prior to antigen binding in order to let the molecule pre-assemble without antigen) is displaced by alterations in the intralysosomal pH.
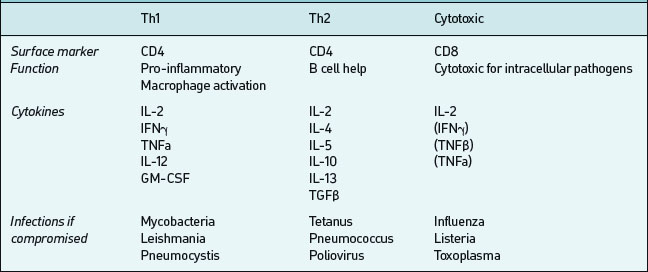
CD4-bearing T cells generally have ‘helper’ functions;those which aid B cell antibody production are called Th2 and those which activate mononuclear phagocytesand promote cellular inflammatory activity are called Th1 (Table 6.5). These T cells types tend to produce different cytokines when activated by antigen; Th1 produce pro-inflammatory IFNγ, IL-1, IL-12, TNFα; Th2 produce IL-4, IL-5, IL-13, TNFα and others which promote antibody production. In any particular immune response one type of T helper activity will often dominate. This is important both in defence against infection and in the pathogenesis of immunologically-mediated diseases. CD8 positive T cells in contrast often have cytotoxic effector properties and are critically important in defense against certain viral infections.
Memory
Once T and B lymphocytes have recognised their cog-nate antigen, they proliferate by producing clones of themselves and acquire effector functions which orchestrate the immune response. Some of these proliferating lymphocytes differentiate into long-lived cells which are called memory cells. B cells differentiate into long-lived plasma cells and some memory B cells are constantly re-stimulated by long-lasting reservoirs of antigen on the follicular dendritic cells (FDC) in lymph nodes. A secondary (memory) response thus involves the activation of an expanded pre-existing panel of antigen-reactive clones, which have differentiated to produce IgG or IgA rather than IgM, giving a response magnified in both quantity and quality. Previously activated cells have been ‘primed’ and thus are more readily activated by small amounts of antigen on APC, since they require less stimulation through their co-receptors. During an immune response, B cells are selected by competition for antigen on FDC in the lymph nodes; those binding strongly survive, others die through inability to compete for antigen and loss of a survival signal. Thus the antibody response undergoes a process of affinity maturation (each generation of antibodies binds better to antigen). Antigen-experienced T – cells differentiate into central memory (CM) T – cells and are primed to produce enhanced secondary immune responses on re-exposure to same pathogen.
T CELL STIMULATION – SIGNALS 1 TO 3
Gell and Coombs classified immune responses in the 1930s (Table 6.6). This is now of limited clinical usefulness since mixed patterns are always seen, but is useful for the general understanding of immuno-logically mediated diseases.
Table 6.6 Classification of specific immune (hypersensitivity) responses
| Type | Mechanism | Clinical example |
|---|---|---|
| I | IgE-mediated | Allergy |
| II | Antibody against cell surface antigens | |
| III | Immune complex deposition | |
| IV | Cell-mediated immunity |
AUTOIMMUNE DISEASES
An autoimmune disease is one in which an immuno-logical attack directed against self-antigens is primarily responsible for the clinical picture. All autoimmune diseases are disorders of the specific immune response. This definition encompasses diseases which affect multiple systems (also known as non-organ-specific) such as systemic lupus erythematosus (SLE) and those which primarily affect a single organ (organ-specific, e.g. Graves’ disease, myaesthenia gravis (MG), autoimmune Addison’s). The immunological effector mechanisms may include direct cellular or humoral responses toan autoantigen, immune complex deposition or interference with normal function of the target antigen (e.g. anti-acetylcholine receptor antibodies in MG, anti-TSH receptor antibodies in autoimmune thyroid disease). Autoimmune diseases are usually more common in women (probably related to oestrogen), may be associated with infections (ankylosing spondylitis, Reiter’s, insulin-dependent diabetes), and are associated with certain MHC haplotypes (A1, B8, DR3) or antigens (e.g. B27, DR3, DR4, DQ2). In many diseases there is dysregulation of immune responses to multiple autoantigens, with an increased incidence of multiple autoimmune diseases.
SELF-TOLERANCE
Autoimmune disease may occur either by reactivation of anergised cells by encounter with potent APCs in certain circumstances which override their programmed unresponsiveness (e.g. where a strong immune response to another antigen results in bystander help sufficient to activate self-reactive T cells in the vicinity), by cross-reactivity between self- and foreign antigens, or as a result of inherited or acquired defects in molecules important in the control of immune responses and maintenance of anergy (e.g. Fas/FasL deficiency-leading to defective apoptosis). The clinical pheno-types of the autoimmunity probably reflect the predominant effector mechanisms and the organ specificity of the antigen(s) and may result in direct damage or interference with normal function. The identity of many autoantigens is now known (Table 6.7). Some are receptors, some enzymes. Autoimmunity may also occur because of failure of induction of self tolerance in the thymus (e.g. autoimmune regulator protein (AIRE) abnormality leading to auto-immune poly-endocrinopathy, candidiasis, ectodermal dysplasia – (APECED)) which results from an inability of the thymic APC to present self antigens to maturing T cells and, therefore, a failure of deletion of self-reactive T cells.
Table 6.7 Autoantigen specificities and disease
| Disease | Autoantigen | Function |
|---|---|---|
| Pemphigus | Desmoglein 1 & 3 | Intracellular adhesion (desmosomal cadherin) |
| Pemphigoid | BPAg 1 & 2 | Basement membrane adhesion (hemidesmosome) |
| Graves’ disease | TSHR (stimulator) | Hormone receptor |
| Hypothyroidism | TSHR (blocking) | Hormone receptor |
| Myasthenia gravis | AchR | Receptor for neuromuscular transmitter |
| Goodpasture’s | NC domain collagen IV | Basement membrane constituent |
| Addison’s disease | Enzymes involved in steroid hormone metabolism (p450SCC shared with ovary/testis) | |
| Autoimmune CAH | p450IID6, p450IIC9, p450IA2 | Liver microsomal enzymes |
| PBC | E2 subunit of pyruvate dehydrogenase | 2-oxoacid dehydrogenase pathway in mitochondria |
Organ-specific autoimmunity manifests itself by damage or malfunction of a single organ as a result of a specific immune response, usually to multiple antigens or to multiple organs on the basis of shared antigens (e.g. steroid cell antibodies linking Addison’s disease and premature ovarian failure, or the lung and kidney damage of Goodpasture’s syndrome). In some conditions (e.g. myasthenia gravis) humoral responses play a major role in many of the disease manifestations, but are unlikely to occur without cellular responses which may also be important. In other diseases, cellular mechanisms may be the predominant pathogenic response: e.g. extrinsic allergic encephalomyelitis (EAE, a model for multiple sclerosis) to myelin basic protein (MBP) and other intracerebral autoantigens. In systemic autoimmunity such as SLE, pathogenesis is multifactorial and involves multiple unrelated antigens. Humoral and cellular responses to multiple nuclear (nucleosome) and cytoplasmic components are seen, particularly anti-double-stranded DNA antibodies (dsDNA) which can cause an immune complex neph-ritis. Sometimes titres of antibodies or complement levels (reduced by immune complex deposition) reflect disease activity in an individual, but in others they do not. In some diseases, the autoantibodies or lymphocytes are pathogenic in models of disease (e.g. anti-dsDNA antibodies in SLE; anti-GBM antibodies in Goodpasture’s). In other diseases they are not, and may be secondary markers of damage (e.g. many antinuclear antibodies (ANA) in SLE, antithyroid peroxidase antibodies in thyroid malignancy).
MHC ANTIGENS AND AUTOIMMUNITY
MHC antigens are inherited (along with a package of minor antigens) as a haplotype consisting of an HLA-A, -B, -C (Class I); -DR, -DP, -DQ (Class II) allele from each parent (Fig. 6.7). Allogeneic immune responses (against a foreign MHC antigen from the same species) can be generated after transplantation.
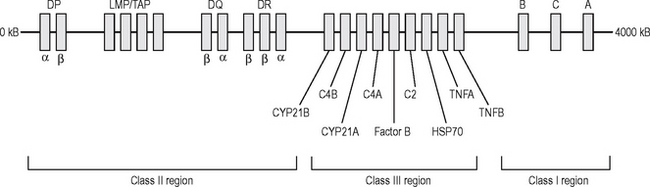
Fig. 6.7 The structure of the human major histocompatibility gene complex (human leucocyte antigen).
The MHC molecule on the APC determines the type and composition of the peptide fragment that it can present to the naïve T cell, and is an important factor in predisposition, protection or disease expression. Certain alleles or haplotypes are associated with particular diseases (Table 6.8). Both organ-specific and non-organ-specific autoimmune diseases are associated with similar MHC haplotypes in some cases, suggesting an inherited predisposition. Few MHC associations with diseases are very strong (most strongly seen between B27 and ankylosing spondylitis), since most conditions are multifactorial and are a result of a combination of genes and additional envir-onmental influences, perhaps including infection.
Table 6.8 MHC associations with disease
| Disease | HLA allele | Relative risk |
|---|---|---|
| Ankylosing spondylitis | B27 | 90 |
| Goodpasture’s syndrome | DR2 | 16 |
| Pemphigus vulgaris | DR4 | 14 |
| Anterior uveitis | B27 | 10 |
| SLE | DR3 | 6 |
| Multiple sclerosis | DR2 | 5 |
| Graves’ disease | DR3 | 4 |
| Rheumatoid arthritis | DR4 | 4 |
| IDDM | DR3 & 4 | 3 |
| Myasthenia gravis | DR3 | 2.5 |
The apparent association of MHC Class I alleles (e.g. HLA-B27) and MHC Class II (e.g. DQB1) may also be due to molecular mimickry between pathogen and MHC, resulting in autoimmune attack. (Heat shock protein (HSP) 60 is widely conserved and generates immune responses in bacterial infection and some autoimmune diseases.). In contrast, some of MHC haplotypes may actually confer protection from some infections and autoimmune disease.
TRANSPLANTATION
TRANSPLANTATION BARRIER
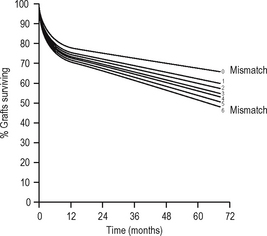
The aim of immunosuppression is to depress the effector immune response to prevent graft rejection (at least initially). The hope is that subsequently either tolerance or graft acceptance will result from downregulation of the antigraft response and enable withdrawal of immunosuppression. The aim of ABO-matching andHLA-matching (tissue typing) is to reduce the antigenic disparity between the graft and the recipient. Other antigens clearly exist (e.g. endothelial antigens) but matching for these is not currently practicable; however, genetic linkage of genes means that related donors with a haplotype match are likely to share the same non-MHC genes. Cyclosporin A (CsA), a fungal metabolite, prolonged survival of renal transplants in man in the late 1970s. By this time, however, graft survival from living related donors had reached a plateau, suggesting that early graft survival results from ABO matching and immunosuppressive drugs, with some contribution from HLA-DR matching (which is more effective than HLA-B or HLA-A matching). Some have, therefore, argued that the benefit of HLA matching is insignificant with modern immunosuppressive drug regimes; however, it appears that long-term graft survival appears more dependent on HLA-A and -B matching (Fig. 6.8).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


