Pathway
Genes
Frequency (%)
MAPK pathway
KRAS
40–45
NRAS
2.5
BRAF
5–10
PI3K pathway
PIK3CA
15
PTEN
10–20
AKT
5
Table 31.2
Overall RAS mutations in CRCs without mutations in KRAS codons 12 and 13
OPUS trial (% of total CRCs) | CRYSTAL trial (% of total CRCs) | |
|---|---|---|
CRCs with other RAS mutations (WT KRAS C12 and C13) | 27.9 | 17.0 |
Other KRAS mutations | 15.2 | 8.9 |
Exon 3: C59 and C61 | 5.9 | 3.3 |
Exon 4: C117 and C146 | 9.3 | 5.6 |
NRAS mutations | 12.7 | 8.1 |
Exon 2: C12 and C13 | 6.8 | 3.5 |
Exon 3: C59 and C61 | 5.1 | 2.8 |
Exon 4: C117 and C146 | 0.8 | 0.9 |
The most frequent BRAF mutations in CRC occur in exon 15 with a T-to-A transversion at nucleotide position 1,796, which leads to the substitution of valine for glutamate (V600E) [16]. The BRAF V600E mutation occurs in 4–12 % of MMR-proficient (microsatellite stable) CRCs and in 40–74 % of MLH1-deficient/MSI-H sporadic CRCs, but is not found in MLH1-deficient MSI-H CRC in HNPCC CRCs [17, 22, 23]. Therefore, determination of BRAF V600E mutation status may differentiate sporadic from hereditary MSI-H CRC, since hereditary CRC will not have a BRAF V600E mutation.
Mutations in the PI3K pathway have been reported in approximately 20 % of all CRCs [16, 17]. Interestingly, mutations involving genes in the two EGFR signaling pathways are not mutually exclusive, and about 5–10 % of CRCs carry mutations in genes from both the MAPK and PIK3 signaling pathways [16, 17].
Cetuximab and panitumumab are antibodies that bind to the extracellular domain of EGFR, block EGF and binding of other endogenous ligands to the EGFR, and thereby block EGFR signaling. Targeted EGFR therapies improve progression-free and overall survival in metastatic CRC [24]. For patients with metastatic CRC with wild-type KRAS, treatment with cetuximab significantly improved overall survival (median 9.5 vs 4.8 months) as compared to supportive care alone (Karapetis et al. [25]). In contrast, patients with mutated KRAS CRCs showed no significant difference between those who were treated with cetuximab and those who were not, leading to the conclusion that patients with CRC carrying a mutated KRAS do not benefit from cetuximab, whereas patients with wild-type KRAS CRC benefit from cetuximab therapy because the antibody effectively blocks the activating EGFR signaling at the cell surface [25]. The mutations tested for in the first trials were limited to KRAS exons 12 and 13 [24]. Further evidence from phase II and III clinical trials using monoclonal antibodies as monotherapy or in combination with chemotherapy for metastatic CRC (stage IV: any T, any N, M1) demonstrated that CRC with a KRAS mutation in codons 12, 13, or 61 did not benefit from treatment with cetuximab or panitumumab [26]. Based on the available clinical trial data in 2009, the American Society of Clinical Oncology recommended that patients with metastatic CRC who are candidates for anti-EGFR antibody therapy should have their tumor tested for KRAS mutations in a CLIA-accredited laboratory [26].
Up to 40 % of patients with a CRC without a KRAS mutation respond to anti-EGFR antibody therapy, while the remaining 60 % of patients with KRAS wild-type tumors did not respond [26]. The lack of response may be due to (1) mutations in genes downstream of KRAS, (2) the presence of KRAS mutations in exons not tested in the assay used for KRAS mutation testing, (3) mutations in NRAS, or (4) changes in other oncogenic pathways in individual cancers. For CRC with mutant KRAS, a number of drugs targeted to inhibit downstream signaling molecules, such as inhibitors of mTOR, RAF, and MEK, are under evaluation [27].
The role of BRAF mutation for prediction of response to anti-EGFR therapy in CRC is still pending results from larger studies [28, 29]. One study reported that colon cancer cells were unresponsive to the BRAF inhibitor vemurafenib due to rapid feedback activation of EGFR, blunting the benefit of vemurafenib [30]. However, the low proportion of CRC cases with BRAF mutations and the poor prognosis associated with BRAF mutations make determination of a definitive impact on outcomes difficult [28, 29].
Conflicting roles of PIK3CA mutations in tumor response to anti-EGFR antibody therapy have been reported in a number of small studies [31, 32]. However, a large study found that the presence of a PIK3CA mutation is associated with a poor response to cetuximab [17]. In this study, BRAF, NRAS, and PIK3CA exon 20 mutations were significantly associated with a poor response to cetuximab, and objective response rates were improved by stratifying patients by genotype of BRAF, NRAS, and PIK3CA exon 20 mutations in the KRAS wild-type population and excluding patients with any mutation from the treatment arm [17, 33]. However, guidelines for testing CRC of patients who are candidates for anti-EGFR antibody therapy for BRAF and PIK3CA mutations have not yet been reported.
Tissue Sample and Gene Testing Considerations for CRC-Targeted Therapies
The standard of practice for selection of patients with metastatic CRC who are candidates for targeted therapies with anti-EGFR antibodies has been primarily based on the mutation status of KRAS [26]. However, as discussed above, the mutation status of BRAF, NRAS, PI3KCA, and other genes downstream of EGFR may affect the tumor response to anti-EGFR therapy [33]. Therefore, testing for mutations in other genes may be indicated in candidate patients, particularly in the setting of clinical trials, at the present time. EGFR mutation testing is not indicated for CRC since activating EGFR mutations in CRC are rare and were not demonstrated to confer sensitivity to tyrosine kinase inhibitors or to cetuximab therapy [34].
In addition to KRAS mutations in codons 12 and 13 of exon 2, KRAS mutations in codons 59 and 61 of exon 3 and codons 117 and 146 of exon 4 and similar mutation hot spots in NRAS need to be considered when selecting true RAS wild-type patients who will benefit from cetuximab therapy for metastatic CRC (Tables 31.2 and 31.3) [20, 21, 35]. Further, the addition of cetuximab therapy for patients with any RAS mutation is of no benefit [20, 21, 35]. Therefore, patients with metastatic CRC should undergo expanded testing for all known activating RAS mutations, to avoid use of cetuximab in patients whose CRCs will not respond [20, 21, 35]. Interestingly, in contrast to other activating mutations in KRAS, use of cetuximab for patients with chemotherapy-refractory CRC with the KRAS G13D mutation may be associated with longer overall and progression-free survival than with other KRAS activating mutations [18], although this has not been supported by some studies.
Clinical trial | ORR (%) | PFS (months) | OS (months) |
|---|---|---|---|
OPUS | 57.9 % vs 28.6 % | 12 vs 5.8 | 19.8 vs 17.8 |
p = 0.008 | p = 0.062 | p = NS | |
CRYSTAL | 66.3 % vs 38.6 % | 11.4 vs 8.4 | 28.4 vs 20.2 |
p < 0.0001 | p = 0.0002 | p = 0.0024 |
Regarding the choice of tissue for DNA mutation analysis, since KRAS mutations occur early in colorectal carcinogenesis, most clinical trials have tested the primary tumor. Published studies show a good correlation between KRAS mutation status in primary and metastatic CRC lesions with an average concordance of 93 % (76–100 %) [36, 37]. Therefore, testing tumor tissue from either the primary site or from a metastatic lesion is appropriate, and a metastatic lesion when available is preferred. Pathologists should select a block of formalin-fixed, paraffin-embedded (FFPE) tissue with the highest proportion of viable tumor and highest percentage of tumor cells as possible, understanding that all cells present in the tissue contribute DNA, so this percentage is of all cells, namely, those that normally compose the colonic wall and variable amounts of inflammatory cells. For small samples, adequate DNA amount can be obtained by pooling macro- or microdissected tumor tissue from multiple tissue levels. This procedure may also provide a larger sampling of the tumor, including areas of mutational heterogeneity, thereby improving mutation detection. Importantly, a pretreatment biopsy sample may be preferable for testing rather than a resection specimen removed after neoadjuvant therapy, as is the case in rectal cancers, where minimal numbers of residual viable tumor cells may persist, making these tissues inadequate for molecular testing. Cytology specimens, particularly from metastatic lesions, also provide an adequate source of tumor for DNA extraction and mutation analysis.
In summary, RAS mutation analysis of CRC tumor tissues is recommended as the standard of care in patients who are candidates for anti-EGFR antibody therapy [20, 21, 35]. Additional mutation testing of other EGFR signaling pathway genes may be helpful to better select patients for targeted therapies with improved outcomes, but a general consensus about which genes should be tested is not yet established. In large practice centers, the trend is to test all CRCs for RAS and BRAF V600E mutations and for MMR deficiency (with the use of immunohistochemical and/or MSI testing), thus allowing for selection of patients for conventional therapy as well as targeted therapy [38]. The use of gene panels for mutation analysis with platforms that permit detection of hundreds of mutations in a single sample, such as next-generation sequencing (NGS) gene panels, may provide further insight into the issue of which genes should be tested. However, determination of which mutations should be considered for CRC treatment selection or exclusion still needs to be better defined based on data from clinical trials. The practice of Genomic Tumor Boards to address the use of gene mutation results and other molecular test results is evolving and is being used in some centers to evaluate treatment options other than established treatment protocols by providing clinical trial options for individual patients.
DNA Mismatch Repair Defects and Microsatellite Instability
Approximately 15 % of all CRCs have a deficient MMR system and are characterized by MSI. In 3–5 % of MMR-deficient/MSI-positive CRCs, patients have germline mutations in MMR genes diagnostic of HNPCC or Lynch syndrome, while the remaining are sporadic-type CRCs [39, 40]. In cells with deficient MMR, errors in DNA replication accumulate and are detectable in short tandem repeats of microsatellite regions but also in functional gene coding regions, which results in changes in the length of repeats at any one location (known as MSI) [41–43]. MSI, specifically when a tumor is identified to have a high level of MSI (MSI-H), is a genomic marker of deficient DNA MMR [43]. Six different genes (MSH2, MLH1, PMS1, PMS2, MSH6, and MLH3) encode the MMR system [44]. In HNPCC, as in other hereditary cancer syndromes, recessive mutation of one allele followed by somatic inactivation of the other is the main mechanism of gene silencing [45]. In contrast, in sporadic CRCs, the most frequent mechanism of DNA MMR gene downregulation is biallelic inactivation by CpG methylation and transcriptional silencing of the MLH1 promoter region [46, 47].
MSI status of CRC can be tested by immunohistochemistry (IHC) of CRC tissue sections to assess expression of the MMR proteins or by PCR-based DNA testing for MSI to detect instability at microsatellite sequences [43, 48]. One advantage of IHC is that the protein identified as showing lost or reduced expression in tumor cell nuclei can be an indicator of which MMR gene has a mutation, which is particularly useful in the workup of HNPCC patients, helping to focus the germline mutation analysis on the most likely affected MMR gene. In addition, combining testing for the BRAF V600E activating mutation and CpG island methylation status of the promoter region of MLH1 can be done to help determine whether a MSI-positive CRC with loss of MLH1 expression is likely to be an inherited HNPCC CRC (BRAF mutation negative and MLH1 promoter methylation negative) or sporadic-type CRC (BRAF mutation positive in up to 70 % of cases and MLH1 promoter methylation positive) [49].
MSI positivity in CRCs is a prognostic marker for improved survival [42]. MSI was detected in 17 % of CRCs of patients < 50 years of age and was associated with (1) a lower likelihood of tumor metastasis to regional lymph nodes as well as distant organs, contributing to improved survival through tumor downstaging, and (2) an overall survival advantage independent of stage of disease [50]. In addition, no significant difference in survival was found between patients with MSI due to hereditary (HNPCC) CRC compared to sporadic CRC [50, 51]. A reported survival benefit for patients with HNPCC CRC is mainly determined by a diagnosis at younger age and less advanced tumor stage as compared with sporadic MSI-H CRC patients [51].
Deficient DNA MMR and MSI status are predictive of response to therapy in some subsets of deficient MMR CRCs [52]. Both the tumor stage and whether the CRC is sporadic or hereditary (HNPCC) are important factors for selection of adjuvant therapy for CRC patients. Patients with stage II CRCs with MMR-deficient/MSI-positive status receiving 5-FU have no improvement in disease-free survival, and treatment is associated with reduced overall survival [53]. The effect of deficient MMR/MSI in stage III CRC on response to therapy has been more difficult to establish, which may be related to heterogeneity of the studied populations in terms of the proportions of sporadic and HNPCC cases. Some studies showed improved outcomes with chemotherapy for advanced stage III CRCs that were MSI-H [54], but other studies did not [55]. In patients with stages II and III CRC, MMR-deficient CRC patients receiving 5-FU had no improvement in disease-free survival [53]. Sinicrope et al. reported that distant recurrences were reduced by 5-FU-based adjuvant treatment in MMR-deficient stage III CRCs, and a subset analysis suggested that any treatment benefit was restricted to suspected inherited CRCs as compared to sporadic tumors [56]. In summary, larger trials are needed to determine with certainty the utility of MMR and MSI status for treatment selection in routine patient care [56, 57]. Current data support the concept that MMR-deficient/MSI-H stage II and III CRC patients do not benefit from 5-FU-based adjuvant therapy, although one study of stage III MMR-deficient CRC patients showed a treatment benefit restricted to patients with suspected hereditary (HNPCC) CRC.
Assays for CRC Mutation Testing for Selection of Therapy
Many technical approaches and assays are used for mutation testing by clinical molecular laboratories. Laboratory-developed tests (LDTs) are frequently used and follow requirements for validation and interpretation as dictated by CLIA and state regulations, while some commercial in vitro diagnostic (IVD) test kits also are used. Assay methods for mutation detection include Sanger sequencing, pyrosequencing, multiplex PCR with primer extension, fluorescent bead detection assays, MassARRAY matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry, allele-specific PCR such as amplification refractory mutation system/scorpions (ARMS/S), melt curve analysis (as performed in Taq-Man assays using real-time quantitative PCR (qPCR)), and, most recently, NGS [58–62]. Table 31.4 compares three technologies commonly used for mutation detection in cancer-related genes in CRC. Until a few years ago, Sanger sequencing of PCR products was a commonly used method for mutation detection. Recent studies comparing Sanger sequencing with other approaches have highlighted its limited analytical sensitivity for clinical testing of DNA from solid tumor samples, requiring 20–50 % mutant alleles in the amplified products [62, 63].
Table 31.4
Comparison of three technologies commonly used for mutation testing for colorectal cancer
Platform | Sanger sequencing | Pyrosequencing | Next-generation sequencinga |
|---|---|---|---|
Principle of technology | Synthesis by addition of labeled nucleotide | Synthesis by nucleotide followed by release of pyrophosphate | Semiconductor: synthesis by nucleotide followed by release of proton |
Detection method | Single sequence of varying length by high-resolution (capillary) electrophoresis | Single sequence of limited length using pyrosequencer | Multiple sequences of varying length using chip-based technology |
Amplification | Incorporation of labeled nucleotides for strand elongation | Incorporation of unlabeled nucleotides followed by light emission | Incorporation of unlabeled nucleotides followed by pH change |
Enzymes | DNA polymerase | DNA polymerase, ATP sulfurylase, luciferase, and apyrase | DNA polymerase |
Advantages | Successfully call repeats. Good visual discrimination for interpretation | Fast. Accurate for short sequence reads. Good visual discrimination for easy interpretation | Emulsion PCR and indexing/bar coding allows for high-throughput multiplexing |
Disadvantages | Low throughput | Relatively low throughput. Need to input sequence. Difficult to read repeats and homopolymers | Labor-intensive. Difficult to read repeats and homopolymers |
Cost | +++ Reagents are expensive | ++ Slightly less expensive than Sanger sequencing | + Comparatively less expensive than both |
Turnaround time | 8 h/gene sequence | 6 h for up to 4 gene sequences | 24–36 h for up to 16 gene sequences |
Technical issues | ++ | + | +++ |
Sensitivity | + | ++ | ++ |
A number of commercial IVD and LDT assays are available to detect the most common mutations in KRAS and BRAF. Many of the early assays only tested for a small number of mutations, limiting the correlation between mutation status and drug response in early clinical trials. Of note, early assays for KRAS mutation analysis performed both in clinical laboratories and as part of clinical trials were limited to codons 12 and 13 in exon 2, excluding other mutation hot spots. Clinical trials have demonstrated the importance of mutations in previously excluded codons of KRAS, in exons 2, 3, and 4, as well as the corresponding codons in NRAS. Additionally, mutations in other genes downstream of EGFR in the EGFR signaling pathway also are clinically significant for patient management decisions, as discussed above.
For mutation testing, genomic DNA can be extracted from fresh, frozen, or FFPE tissue using standard molecular laboratory methods. For solid tumor testing, histologic slides should be reviewed by an anatomic pathologist to assess the percent of tumor cells relative to total cells on the slide and to prioritize areas for macro- or microdissection, if necessary, to increase the percentage of tumor cells in the sample used for testing.
Pyrosequencing Assays
Pyrosequencing is a DNA sequencing technique based on the sequencing-by-synthesis principle [64], which is used for analysis of gene regions that are frequently mutated. A pyrosequencing result (pyrogram) for BRAF exon 15 is shown in Fig. 31.1. Non-mutated samples show the wild-type sequence with appropriate peaks and peak heights. Mutations are indicated as different peaks compared to the wild-type sequence or alterations in the height of wild-type peaks relative to a neighboring peak or compared to the reference peak. A reference peak is usually the first peak on each pyrogram. If the background is too high or the peaks are too low, pyrosequencing should be repeated. Indeterminate cases can be verified by Sanger sequencing or other available assays. Examples of BRAF mutation testing by pyrosequencing from CRCs of two different patients are shown in Fig. 31.2.
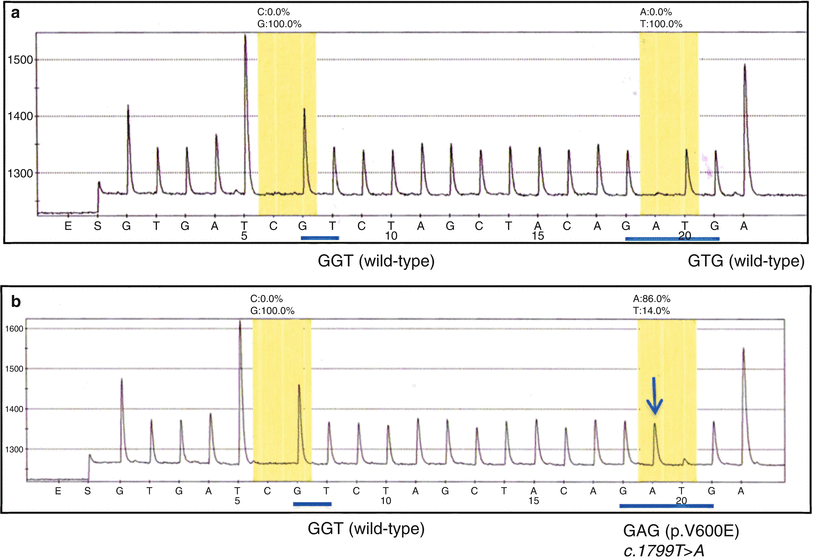
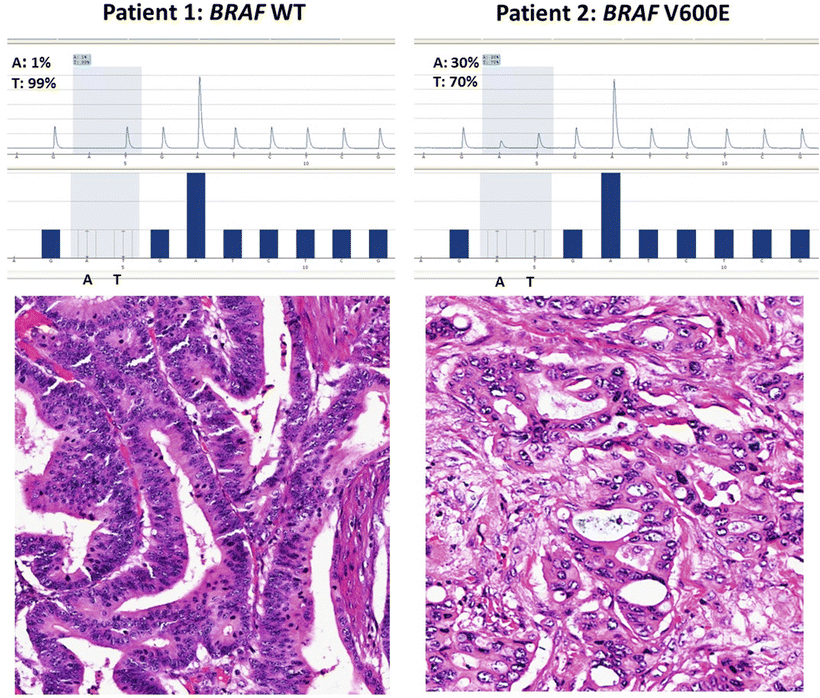
Figure 31.1
Pyrosequencing analysis of BRAF exon 15. (a) Wild-type BRAF sequence. (b) An “A” peak (downward blue arrow) is present instead of the “T” peak in the wild-type BRAF sequence, due to a T>A mutation (c.1799T>A), which results in an amino acid change from valine to glutamic acid in the BRAF protein (p.V600E)
Figure 31.2
Colorectal cancers from two patients tested for BRAF V600E mutation by pyrosequencing. Patient 1 (left) has a wild-type (WT) gene (99 % T allele at position 1,799) and patient 2 (right) is positive for the BRAF V600E mutation (30 % A allele at position 1,799). Note that the “A” allele is only present at 30 % of total alleles due to DNA from non-tumor cells in the tissue section that have only the wild-type T allele
Compared to Sanger sequencing, which has an analytic sensitivity of 20 % mutant alleles, pyrosequencing has a higher sensitivity of approximately 5 % using FFPE tissue [65–69]. With its short read length (approximately 50 base pairs), pyrosequencing is optimal for scanning for mutations in hot spot gene regions and has been used to detect mutations in KRAS codons 12, 13, 61, and 146 and the BRAF V600E mutation [70–72]. Pyrosequencing is suitable for single-nucleotide polymorphism (SNP) genotyping [73] and DNA methylation analysis [74, 75], such as MLH1 methylation in CRC [76, 77]. The assay can be performed in 96- or 384-well plates and scaled for sequencing hundreds of megabases of DNA using a large-scale parallel pyrosequencing system.
The limitations of pyrosequencing include the short read length of DNA sequences and sequencing errors in homopolymer regions, since the light intensity in such regions is not proportional to the number of nucleotides incorporated.
Multiplexed Primer Extension Assays
The primer extension method is a template-directed dye termination assay designed to detect the base immediately 3′ to the sequencing primer [78]. After the sequence of interest is amplified by PCR and the unincorporated nucleotides are removed, primer extension can be performed in a single reaction tube using, for example, the SNaPshot Multiplex kit (Applied Biosystems, Foster City, CA). The four major steps in a primer extension assay are (1) PCR amplification and column purification of the PCR product; (2) primer extension; (3) alkaline phosphatase digestion and heat inactivation; and (4) capillary electrophoresis of the fluorescently labeled products. Concurrent detection of mutations at multiple sites is possible by multiplexing at the PCR step [79]. When multiplex reactions are run, the primer extension probes are designed with varying lengths of deoxythymidine monophosphate homopolymers at their 5′ end to allow for discrimination of the probe for each site of interrogation by size.
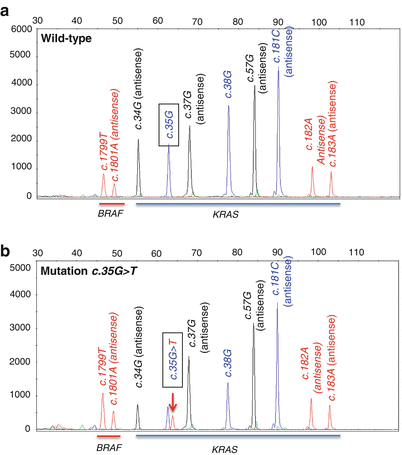
An example of a multiplex primer extension assay for concurrent detection of BRAF V600 and K601 and KRAS exons 2 and 3 mutations in CRC is shown in Fig. 31.3. Genomic DNA from FFPE CRC tissue is isolated, and the coding sequences for BRAF exon 15 and KRAS codons 12, 13, and 61 are amplified using specific primers in a multiplex PCR reaction. The probes for primer extension are either in the sense or antisense direction and end one base 5′ of the following positions: nucleotides 1,799 and 1,801 of the BRAF gene and nucleotides 34, 35, 37, 38, 181, 182, and 183 of the KRAS gene. The probes allow for detection of common variants present in codons V600 and K601 of the BRAF gene and codons 12, 13, and 61 of the KRAS gene. After amplification, the PCR products are incubated with the unlabeled oligonucleotide primer extension probes, four dideoxynucleotide triphosphates (ddNTPs) labeled with different fluorescent colors, and DNA polymerase. During this primer extension step, only the next complementary 5′ nucleotide is added to each probe. The primer extension products are analyzed by capillary electrophoresis. The base at the mutation site is determined by detection of the fluorescent color of the ddNTP that is incorporated.