Figure 15.1
The Coagulation Cascade: a activated factor; Ca 2+ calcium; PL phospholipid. Reprinted with permission from Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance and regulation. Biochemistry. 1991;30:10363. Copyright 1991 American Chemical Society
Hemophilia A
Overview
HA is an X-linked recessive bleeding disorder due to a deficiency in coagulation factor VIII (FVIII). HA affects approximately 1 in 10,000 live male births, among all ethnic populations. Approximately 30 % of cases result from new mutations occurring in families in whom there is no apparent family history of HA. The diagnosis of HA is established based on reduced or absent FVIII activity (FVIII:C) and is classified into severe (<1 %), moderate (1–5 %), or mild (>5–40 %) disease, with estimated prevalence of 43 %, 26 %, and 31 %, respectively [1].
Molecular Basis of Disease
The factor VIII gene (F8) is located on the long arm of the X chromosome (Xq28) (Fig. 15.2). F8 is 186 kilobase pairs (kbp) with 26 relatively short exons, ranging from 69 to 262 base pairs (bp), and two long exons, exon 14 (3,106 bp) and exon 26 (958 bp). The resulting messenger RNA (mRNA) is approximately 9 kb, of which the coding sequence is 7,053 nucleotides. The intron/exon boundaries roughly correlate with the FVIII domains (Fig. 15.3). The introns are large (14–23 kb) with intron 22 being the largest (32 kb). A CpG island in intron 22 acts as a bi-directional promoter for two additional genes (Fig. 15.2). The first, termed F8-associated gene A (F8A or Int22h-1), is an intronless gene approximately 2 kb long and is transcribed in the opposite direction to F8. The second F8-associated gene B (F8B) is 2.5 kb long and transcribed in the same direction as F8. The F8A and F8B transcripts originate within 122 bases of each other and the functions of their potential protein products are unknown. The F8A sequence is replicated at least twice, approximately 500 kb telomeric to the F8 gene and close to the tip of the X chromosome termed Int22h-2 (proximal) and Int22h-3 (distal). Int 22h-2 and -3 are about 100 kb apart and transcribed in the same direction as the F8 gene. These three homologous repeats are involved in the intron 22 inversion mutation which is a frequent rearrangement of the F8 gene, resulting in severe HA (Fig. 15.4). The putative promoter region is located 300 nucleotides 5′ to the gene and although a TATA box is not essential for transcription, liver-enriched transcription factors (e.g., HNF1, NFkB, C/EBPa, and C/EBPb) interact with the F8 promoter region. The F8 gene encodes a precursor protein of 2,351 amino acid residues consisting of a 19 amino acid leader peptide followed by 2,332 amino acids in the mature protein. The mature protein can be divided into several homologous domains termed A1-A2-B-A3-C1-C2 and is predominantly expressed in the liver.
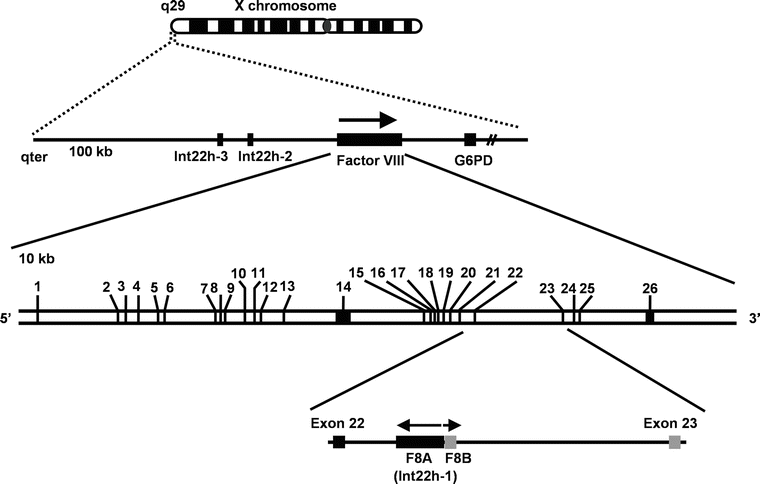
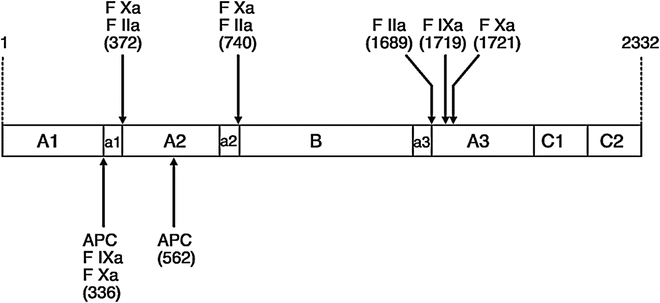
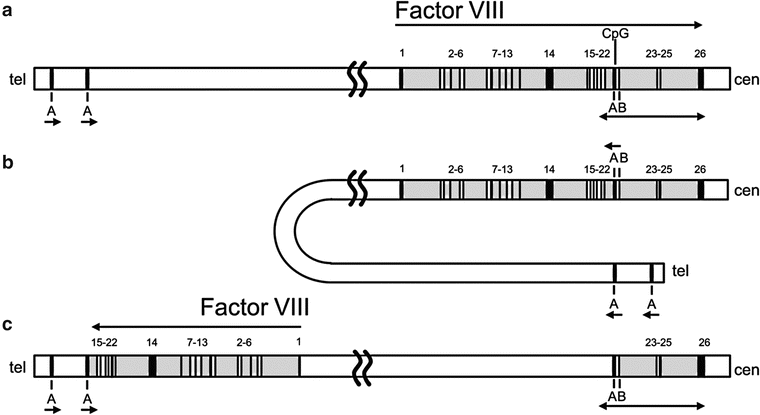
Figure 15.2
The location and structure of the F8 gene: G6PD glucose 6 phosphate dehydrogenase, kb kilobases. Reprinted with permission from Kazazian HH, Tuddenham EGD, Stylianos EA. Hemophilia A: deficiency of coagulation factor VIII. In Scriver CR, Beaudet al, Valle D, et al., eds. The Metabolic and Molecular Basis of Inherited Disease. Copyright 2001 McGraw-Hill
Figure 15.3
FVIII peptide showing domains A1 to C2, cleavage sites (arrows) for thrombin (FIIa), activated factor X (FXa), and activated protein C (APC): Amino acids are numbered in parentheses. The heavy-chain cleavage site (A1-a1-A2-a2) is linked to the light-chain cleavage site (a3-A3-C1-C2) via the B domain. From Pruthi RK, Nichols WL. Autoimmune factor VIII inhibitors. Curr Opin Hematol 6;314. Copyright 1999 Lippincott Williams and Wilkins. Reprinted with permission from Wolters Kluwer Health
Figure 15.4
Intron 22 inversion mutation of the F8 gene: (a) Orientation of the F8 gene showing three copies of the F8A gene (A) and location of the F8B gene (B) transcript in intron 22. Arrows indicate the direction of transcription of the F8 and internal F8A (A) and F8B (B) genes. (b) Proposed mechanism of homologous recombination between intron 22 gene F8A (A) and one of the two telomeric copies of gene F8A (A). (c) Results of proposed crossover. cen centromere, tel telomere. Reprinted with permission from Kazazian HH, Tuddenham EGD, Stylianos EA. Hemophilia A: deficiency of coagulation factor VIII. In Scriver CR, Beaudet al, Valle D, et al., eds. The metabolic and molecular basis of inherited disease. Copyright 2001 McGraw-Hill
Factor VIII is activated to FVIIIa via proteolytic cleavage, by either thrombin (FIIa) or factor Xa in the presence of phospholipid surfaces (Fig. 15.3). Although cleavages at amino acid positions 740 or 1,721 have no effect on coagulant activity, the cleavages at amino acid position 372 or 1,689 are important for FVIII procoagulant activity. Cleavage at 1,689 releases FVIII from VWF, permitting FVIII interaction with phospholipids and platelets. Missense mutations affecting these cleavage sites have been found in patients with HA and result in a form of hemophilia called “cross-reacting material positive (CRM+)-HA” with normal levels of FVIII antigen but low activity (1–7 %). Although missense mutations have been found in the A2 domain in HA and its value to FVIII coagulant activity has been confirmed by in vitro studies, the exact role of the A2 subunit remains unknown.
The B domain is cleaved during proteolytic activation and is not directly required for procoagulant activity of FVIII. However, it may have a role in intracellular processing and/or secretion of FVIII as B-domain-deleted FVIII molecules are expressed at five- to ten-fold higher levels than non B-domain-deleted FVIII. Mutations in this region have been reported in HA.
VWF-bound FVIII is protected from inactivation by activated protein C (APC). The putative VWF-binding region on FVIII is felt to be at the N-terminus of the light chain of FVIII and in the C2 domain. The binding site for FIXa has been localized to the A2 domain and regions of the light chain. In addition, the binding site to FX is localized to the C-terminus of the A1 domain. Binding to phospholipids, which is important for FX activation by FIXa and FVIIIa, occurs in the C1 and C2 domains of the light-chain FVIII. No deleterious mutations in HA patients have been identified in the inactivation cleavage sites of FVIII.
Deleterious Mutations in F8 Gene
Deleterious mutations and polymorphisms are cataloged in an international database available on the Internet and updated periodically: http://hadb.org.uk. References for the amino acid numbering system used below can be accessed at this website.
Mutations have been found in the promoter and all coding regions of the F8 gene. Of the approximately 1,492 unique reported mutations, missense mutations account for approximately 46 %, small deletions (<50 bp) 18 %, large deletions (>50 bp) 11 %, nonsense mutations 10 %, and splice-site mutations and insertions 7 % each. However, approximately 40 % of severe HA patients have an inversion mutation at the tip of the X chromosome that disrupts the F8 gene [2]. Homologous recombination can occur when the F8A gene (Inth22-1) in intron 22 of F8 pairs with one of the two homologous regions (Int22h-2 or Int22h-3) that are telomeric to the F8 gene probably as a result of folding over of the tip of the X chromosome (Fig. 15.4). Upon unfolding, exons 1–22 are inverted and placed approximately 500 kb upstream of exons 23–26 and oriented in the opposite direction. Depending on which repeat the F8A pairs with, the inversion may be termed type I (distal) or type II (proximal). Rarely type III inversion mutations occur in patients with a third extragenic copy of F8A. Given that the majority of inversions originate in male meiosis, almost all mothers of patients with the inversion mutation are carriers. An additional inversion of exon 1 of the F8 gene affecting up to 5 % of patients with severe HA has also been described [3].
Genotype-Phenotype Correlations
The types of F8 mutations correlate with the severity of hemophilia and the risk of developing FVIII inhibitors. In addition to the inversion mutations, certain missense, insertion, and deletion mutations result in severe disease (<1 % FVIII activity). However, specific deletions of 156 bp of exon 22 or the 294 bp of exons 23 and 24 are associated with moderate disease, likely due to in-frame splicing of exon 21 to exon 23, or exon 22 to exon 25. Patients with large deletions are susceptible to formation of FVIII inhibitors (antibodies) in response to therapy with FVIII concentrates. In an analysis of the HA database, up to 40 % of patients with deletions develop FVIII inhibitors, whereas up to 60 % of patients with single-base pair changes resulting in nonsense mutations and 15 % of patients with single-base pair changes resulting in missense mutations developed inhibitors [4]. More recently, a meta-analysis of 30 studies involving over 5,000 patients concluded that the pooled odds ratios (OR) of inhibitor development in large deletions and nonsense mutations were 3.6 (95 % CI 2.3–5.7) and 1.4 (95 % CI 1.1–1.8), respectively, compared to intron 22 inversion mutations, with the remaining mutations (e.g., intron 1 inversions and splice-site mutations) posing an equivalent risk as an intron 22 inversion [5].
Single-base pair changes that result in missense mutations are spread throughout the F8 gene. Although the structure-function relationships of some of the missense mutations are known or can be deduced (e.g., alteration of VWF-binding site, thrombin cleavage site), the structural consequences of most such mutations remain undefined.
Although HA predominantly affects males, some female carriers have reduced FVIII:C and may have clinically significant bleeding. The molecular basis of symptomatic females includes lyonization of the normal X chromosome and Turner syndrome (XO karyotype) where the dominant mutant F8 gene is responsible for production of the abnormal FVIII. Rarely, a female known to be a carrier may have children with a known hemophilia patient; in this circumstance, each child regardless of gender has a 50 % chance of being affected.
Polymorphisms in the F8 Gene
Polymorphisms present within the F8 gene (intragenic) or outside the F8 gene (extragenic) have been used to assign haplotypes (combination of polymorphisms) for linkage analysis. The putative defective F8 gene can be tracked with polymorphisms that are closely linked to the gene. The carrier frequency of such polymorphisms varies with the ethnicity of the study population and needs to be considered when studying patients of diverse ethnic origin. A complete listing of polymorphisms is available in the F8 mutation database at http://hadb.org.uk.
Clinical Utility of Testing
In a male patient suspected of being affected, the diagnosis of HA is established by assaying plasma FVIII:C. Given that FVIII relies on binding to VWF for its normal survival, all patients with a low FVIII:C should have VWB excluded. In addition, FVIII is a labile factor resulting in a potential artifactual loss of up to 15 % of FVIII:C with delayed processing of plasma specimens. Thus, mild reductions in FVIII:C should prompt repeat testing with careful attention to timely specimen processing.
In at-risk females, while reduced FVIII:C typically confirms carrier status, a normal FVIII:C does not exclude the possibility of carrier status. Given that FVIII:C may be normal in carriers and the fact that FVIII is an acute-phase reactant, it can be elevated with acute or chronic inflammation, liver disease, vasculitis, estrogen therapy, or pregnancy. Thus, molecular genetic testing would be the only option for diagnosis.
Knowledge of the causative F8 mutation in the proband does not alter clinical management but may be useful in predicting the risk of developing FVIII inhibitors [4]. The causative mutation can be useful for carrier testing of family members. Asymptomatic females with no access to their familial F8 genotype information will need their entire F8 gene analyzed for carrier testing. This is not an uncommon situation for adopted females. For carriers, prenatal diagnosis during pregnancy provides useful information for management of labor and delivery and, occasionally, pregnancy termination. Advances in reproductive technology may permit preimplantation genetic diagnosis and circumvent the need for prenatal testing [6].
Available Assays
Several laboratories offer genetic testing for HA. A complete listing of clinical laboratories performing HA testing that have registered with GeneTests can be found on their website (www.genetests.org). An alternative source is the NCBI Genetic Testing Registry (http://www.ncbi.nim.nih.gov/gtr). In general, testing can be divided into direct mutation analysis and indirect testing based on haplotype, and selected laboratories offer prenatal testing.
Direct DNA Analysis
Direct DNA analysis for the intron 22 and 1 inversion mutations is typically performed by restriction digestion and Southern blot method. This detects the more common types: I and II inversions and the rare type III inversion. Polymerase chain reaction (PCR)-based assays also can be used [3, 7, 8]. Detection of hemizygous gene or exonic deletions in the F8 gene in at-risk carriers by PCR amplification of individual exons may not be diagnostic given that the normal copy of the gene may be amplified. For this situation, linkage analysis may be required; however, the role of multiplex ligation-dependent probe amplification (MLPA) is evolving [9]. Different laboratories utilize varied approaches, including initial screening or direct sequencing of all the relevant regions of the F8 gene [10]. The large size of the F8 gene with 26 exons makes this a labor-intensive test. Thus alternate strategies have been utilized on a research basis which involves analysis of F8 mRNA by RT-PCR [11] or other SSCP-based screening techniques [12].
Indirect DNA Analysis (Linkage Analysis)
The principle underlying genetic linkage analysis is the tendency for alleles close together on the same chromosome to be transmitted together as an intact unit through meiosis. Polymorphisms, within the F8 gene (intragenic) or outside the F8 gene (extragenic), are typically single-base pair changes or simple sequence repeats (CAn repeats). Disadvantages of linkage analysis include the need for DNA samples from the proband and both parents, and the requirement that the proband’s mother be heterozygous and thus “informative” for the polymorphism. A polymorphism is informative if the DNA sequence at a locus differs on the maternally inherited and paternally inherited chromosomes. Most families (up to 90 %) are informative with one or more DNA polymorphisms if both intragenic and extragenic polymorphisms are analyzed. The distance between the disease-causing mutation and the polymorphism increases the risk of recombination, which may lead to false-positive or false-negative results. This risk is lowest if intragenic markers are used. Rarely, families will only be informative for extragenic polymorphisms: misdiagnosis can occur in up to 6 % of carriers as a result of linkage disequilibrium. When an intragenic polymorphism is used for the diagnosis of carrier or affected status, the chance of an error is less than 1 %. A major drawback with linkage analysis is the possibility of identifying nonpaternity in the tested family. A complete listing and frequency of polymorphisms is available in the F8 mutation database.
HA Testing Algorithms
HA genetic testing should be accompanied by pretest and posttest counseling, which is described below. For questions regarding any laboratory’s sample requirement, testing, and reporting process, direct communication with the laboratory is critical. Most laboratories have a genetic counselor available for testing guidance. In general, genetic testing for hemophilia is ordered by healthcare providers familiar with the diseases, inheritance patterns, and testing options, but also is ordered by primary care providers who may not be familiar with testing options and counseling. Algorithms for testing symptomatic males and at-risk females are illustrated in Figs. 15.5 and 15.6 and described below.
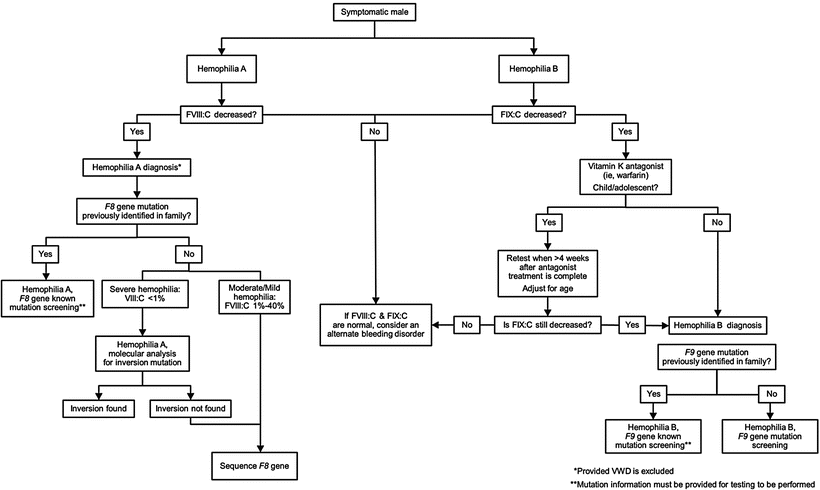
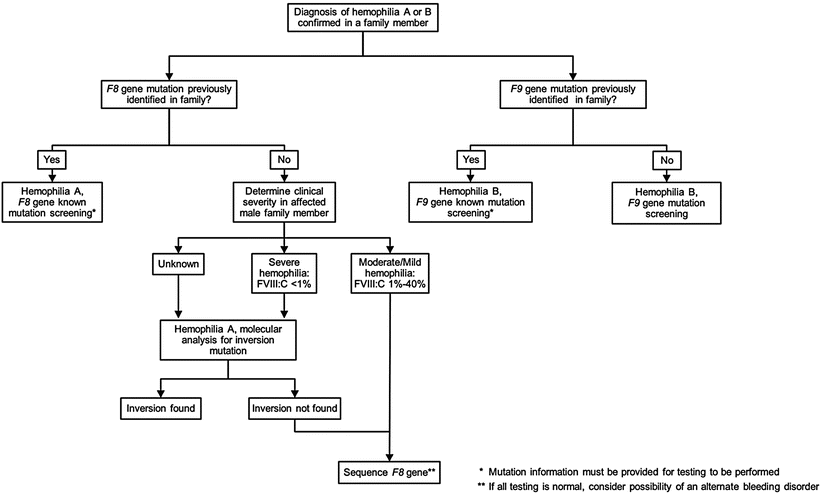
Figure 15.5
Hemophilia testing algorithm: symptomatic male. FVIII:C, FVIII activity; FIX:C, FIX activity; F8, FVIII gene; F9, factor IX gene. By permission of Mayo Foundation for Medical Education and Research. All rights reserved
Figure 15.6
Hemophilia carrier testing algorithm: FVIII:C, FVIII activity; FIX:C, FIX activity; F8, FVIII gene; F9, factor IX gene. By permission of Mayo Foundation for Medical Education and Research. All rights reserved
For a symptomatic male, the diagnosis of HA is established based on plasma FVIII:C, which also classifies the disease as severe (<1 %), moderate (1–5 %), or mild (>5–40 %) (Fig. 15.5). In patients with a history of a bleeding disorder but who have normal FVIII:C, alternative bleeding disorders should be considered, e.g., HB (X-linked recessive) or VWD (autosomal dominant or recessive). If the specific F8 mutation has been identified in an affected family member, the documentation of the specific familial mutation should be provided to the laboratory and testing should concentrate on identifying the presence or absence of the familial mutation; typically, testing laboratories offer focused testing generically termed “known mutation analysis.” If the familial mutation has not been identified, for symptomatic males with severe HA, initial testing is focused on the F8 intron 22 inversion mutation and, if negative, the intron 1 inversion. For those with no detectable inversion mutation and for probands with moderate or mild HA, full gene analysis is required.
For at-risk females, a reduced FVIII:C confirms carrier status; however, a normal FVIII does not exclude HA carrier status, for which molecular testing is required. As with testing male patients, if the familial mutation is known, this information should be provided to the laboratory and testing should focus on the familial mutation (Fig. 15.6). For at-risk females without a known familial mutation, determining the clinical severity of hemophilia in an affected male family member will aid in a rational testing approach. Information on severity of the hemophilia may not be known to the at-risk female (especially if the proband was a distant relative). Communication with the local comprehensive hemophilia center that provided hemophilia care to the proband may be useful. For families with severe HA, initial testing is focused on the F8 intron 22 inversion, and if negative, the intron 1 inversion. For those with no detectable inversion mutation and for families with moderate or mild HA, full F8 gene analysis is required.
Identification of a causative mutation confirms the disease in symptomatic males, and carrier status in at-risk females. This carrier status confers a 50 % risk of each male child being affected with and a 50 % risk of each female child being a carrier for HA. All biological daughters of an affected male are obligate carriers. Lack of a detectable mutation in an at-risk female excludes carrier status if the familial mutation is known, but not if the familial mutation is unknown. Posttest counseling for a discussion on the aforementioned issues is vital in ensuring that the patient understands his or her own risk and the risk for offspring to be carriers of or affected with HA.
Interpretation of Test Results
Reduced FVIII:C is virtually diagnostic of HA, provided VWD and specimen artifact (see above) have been excluded. Rare instances of other genetic disorders that can have low FVIII activity include VWD type 2N (see section on VWD) and rare combined deficiencies of FV and FVIII. The latter occurs as a result of mutations in ERGIC-53, which is a protein necessary for efficient transport of FV and FVIII from the endoplasmic reticulum to the Golgi. FV and FVIII levels are typically in the 10–15 % range [13].
Detection of the presence of a deleterious mutation in a symptomatic male establishes the familial mutation and confirms carrier status in the at-risk female. Certain well-defined mutations (e.g., inversion, deletions, insertions, splice junction mutations, and nonsense mutations) have obviously deleterious effects. Determining the deleterious nature of previously uncharacterized missense mutations poses a challenge. However, identification of a variant of uncertain significance in hemizygous genes (F8 and F9) in males without additional mutations provides reasonable evidence of its deleterious nature. Other criteria typically considered include genotype-phenotype correlation in the family if other family members are affected and tested, analysis of the degree of conservation of the respective residue among other species, and presence of a similar mutation in other patients. The best evidence of deleterious nature of a missense mutation is in vitro confirmation of its effect on protein function.
Laboratory Issues
As with any genetic test performed on peripheral blood leukocyte genomic DNA, high-quality DNA extracted from whole blood is essential. The specimen needs to be clearly identified and, given that testing is performed on peripheral blood leukocyte genomic DNA, history of allogeneic bone marrow or peripheral blood stem cell transplant needs to be provided. For testing performed on cord blood samples, a maternal peripheral blood sample should also be submitted for exclusion of maternal cell contamination of the cord blood which can cause incorrect test results.
Hemophilia B
Overview
HB is an X-linked recessive bleeding disorder due to a deficiency of FIX and is clinically indistinguishable from HA. HB affects 1 in 30,000 live male births across all ethnic groups. As with HA, up to 30 % of HB cases occur in families with no prior family history of HB. The diagnosis is established based on plasma FIX activity (FIX:C), which permits classification of HB disease severity as severe (<1 %), moderate (1–5 %), or mild (>5–40 %).
Molecular Basis of Disease
The factor IX gene (F9) is located on the long arm of the X chromosome (Xq27.1) and is 38 kb long with eight exons of varying lengths (25–1,935 bp). The F9 mRNA is approximately 3 kb in length with a coding sequence of 1,390 nucleotides. The intron/exon boundaries roughly correlate with the FIX domains and bear a high degree of homology to members of the vitamin K-dependent protein family (FVII, FX, and protein C) (Fig. 15.7). The F9 gene encodes a precursor protein of approximately 454 amino acids consisting of a propeptide followed by a glutamic acid-rich (Gla) domain, two epidermal growth factor domains, an activation peptide, and a catalytic domain.
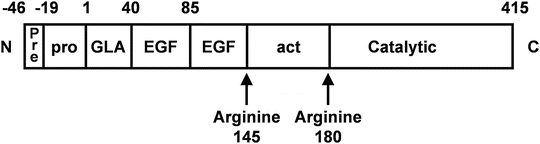
Figure 15.7
Factor IX peptide showing domains: N N-terminus, C C-terminus, Pre pre-propeptide, pro propeptide, GLA glutamic acid-rich domain, EGF epidermal growth factor domain, act, activation peptide
Activation of FIX to FIXa occurs by cleavage by factor VIIa (FVIIa)-tissue factor (TF) and activated factor XI (FXIa). Cleavage releases the activation peptide, resulting in circulating light and heavy chains connected by a disulfide bond. Numerous posttranslational modifications are necessary for the normal function of FIXa, including tyrosine sulfation, serine phosphorylation, and O– and N-linked glycosylation. The relatively small size of the F9 gene allows complete molecular testing, and mutations are catalogued in a database accessed at http://www.factorix.org.
Mutations in the F9 Gene
The majority of F9 mutations are single-base pair changes that result in missense, frameshift, or nonsense mutations. Short deletions (<30 nucleotides) account for approximately 7 %, larger deletions approximately 3 %, and insertions approximately 2 % of mutations. Many of the single-base pair changes occur at CpG doublets that are “hotspots” for mutation. However, a subset of recurrent mutations is due to a founder effect, which typically results in mild disease. Mutations have been detected in all regions of the F9 gene, including the poly(A) signal.
Genotype-Phenotype Correlation
The numbering system for FIX peptide has evolved. The former numbering system was based on the Yoshitake numbering system [14] which is termed the legacy system in the FIX mutation database. An alternate and increasingly used system is the Human Genome Variation Society numbering system. The numbering system utilized in this chapter is the legacy system.
Missense mutations account for the majority of mutations that typically result in mild disease unless the mutations occur in residues critical for normal FIX function. Selected mutations in the promoter region of the F9 gene result in a unique phenotype termed hemophilia B Leyden. This phenotype is characterized by severe disease at birth with progressive amelioration of severity through adolescence and puberty. However, some promoter mutations (e.g., Brandenburg mutation at −26) in the F9 promoter result in lifelong severe disease. Nonsense mutations in the signal peptide and propeptide regions lead to severe HB. Selected missense changes, that lead to retention of FIX within hepatic cells (e.g., p.Ile-30 and p.Ile-19) or prevent cleavage of the propeptide, result in a dysfunctional FIX molecule (e.g., p.Arg-4) and result in hemophilia of varying severity (mild, moderate, or severe) depending on the amino acid substitution.
Mutations in the Gla domain disrupt γ-carboxylation (posttranslational modification) that is important for normal FIXa binding to collagen, activated platelets, and endothelial cells. Mutations in the EGF domains result in disruption of FIX binding to calcium that is essential for procoagulant activity, as well as impaired binding to its cofactor FVIII. Mutations in the catalytic domain typically disrupt the catalytic triad (p.His221, p.Asp269, and p.Ser365) essential for FIXa protease function.
An unusual FIX variant, due to mutation at p.Ala10, is characterized by normal baseline FIX:C. However, warfarin therapy results in a severe and disproportionate reduction in FIX:C (typically <1 %) and causes bleeding in patients being treated with warfarin who have an apparently therapeutic International Normalized Ratio. An indication of such a situation is a disproportionate prolongation of the activated partial thromboplastin time which should prompt testing for FIX:C levels. A second unusual variant, p.Arg338Leu (termed FIX Padua), results in a marked increase in FIX:C of up to 700 % of normal. In one case report, this mutation resulted in early-onset and recurrent VTE [15].
Polymorphisms in the F9 Gene
Eight common polymorphisms that do not cause HB have been described in different ethnic populations of European and African descent. These polymorphisms, however, are much less common in Asian and other populations. The most informative polymorphism documented in the Asian population is what is termed the Hha I polymorphism (rs3117459 allele frequency = 0.17) located approximately 8 kb 3′ to F9. Recent reports [16] have described additional polymorphic loci in these populations, facilitating molecular diagnosis of non-Caucasian carriers and patients with HB. A polymorphism within the FIX-coding region, p.Ala148Thr, occurs within the activation peptide. This does not correlate with FIX activity or antigenic levels. The Thr allele occurs with a frequency of 0.3 in the Caucasian population; however, it is much less frequent in the African-American (0.053–0.15) and Asian (<0.01) populations.
Clinical Utility of Testing
The diagnosis of HB in a symptomatic patient is established by FIX:C testing, rather than by genetic testing. Given that FIX is a vitamin K-dependent protein, all patients with mild-to-moderate reductions in FIX activity should have vitamin K deficiency excluded. In addition, healthy normal children have a lower FIX activity that reaches adult reference ranges at puberty.
For the symptomatic male, knowledge of F9 genotype does not alter clinical management of HB patients; however, the specific genotype may predict the risk of developing FIX inhibitors and anaphylaxis in response to FIX concentrate therapy [17]. Although knowledge of the proband’s genotype information is important for carrier testing of at-risk female family members, the smaller size of the F9 gene allows routine use of sequence analysis to identify the specific F9 mutation causing HB. A reduced FIX:C in at-risk females typically confirms HB carrier status, provided that vitamin K deficiency has been excluded. However, normal FIX:C does not exclude the patient being a carrier. In this circumstance, molecular genetic testing would be the only option for diagnosis. See Fig. 15.5 for a recommended diagnostic testing algorithm for HB patients and Fig. 15.6 for recommended testing algorithm for HB carriers.
Available Assays
A listing of laboratories offering HB genetic testing that have registered with GeneTests can be found on their website (www.genetests.org). An alternative source is the NCBI Genetic Testing Registry (http://www.ncbi.nlm.nih.gov/gtr/). In general, testing can be divided into direct and indirect testing and selected laboratories offer prenatal testing.
Direct DNA Analysis
Generally, most patients with HB have mild disease. Since approximately 25 % of mild HB Caucasian patients have one of the three founder mutations (p.Gly60Ser, p.IIe397Thr, and p.Thr296Met) [18], a logical first step is to perform limited testing for these founder mutations. For symptomatic patients without one of the founder mutations, for severely affected patients, and for individuals at risk of being an HB carrier, the logical next step is F9 gene mutation screening or sequencing regions of functional significance in the F9 gene. Although the majority of HB patients have one deleterious mutation, approximately 1 % of HB patients have two mutations. Additional limitations of molecular testing as described in the section above on HA also apply for HB.
Indirect DNA Analysis (Linkage Analysis)
As discussed above for HA, indirect DNA analysis of an HB proband does not identify the familial mutation. Linkage analysis does assign a haplotype which identifies the abnormal F9 gene and is useful for carrier testing of family members. Given the small size of the F9 gene, direct sequencing is also feasible for carrier testing when the HB mutation is not a large F9 gene deletion. As discussed in the section on HA, such mutations may be detected by MLPA.
Testing Algorithm
Pre- and posttest genetic counseling is critical for overall patient management. See section on HA above. A reduced, age-appropriate, FIX:C confirms the diagnosis of HB, provided that vitamin K deficiency or use of vitamin K antagonists (e.g., warfarin) can be excluded. The FIX:C level allows classification of the severity of disease as severe (<1 %), moderate (1–5 %), or mild (>5–40 %). If FIX:C is not reduced, consider testing for an alternate bleeding disorder, such as HA or VWD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


