Cholinergic pharmacology is centered on the properties of the first identified neurotransmitter, acetylcholine (ACh). The functions of cholinergic pathways are diverse; generally, they involve the neuromuscular junction (NMJ), the autonomic nervous system, the central nervous system (CNS), and the non-neuronal cholinergic system (NNCS). In the neuronal cholinergic systems, ACh acts as a neurotransmitter at the NMJ, at autonomic ganglia, at terminal synapses of parasympathetic postganglionic fibers and a few sympathetic postganglionic fibers, and in the CNS. Many non-neuronal cells also express ACh receptors and can thereby serve as effector cells for both neuronally and non-neuronally released ACh.
Despite the many physiologic actions of ACh, the current therapeutic uses for cholinergic and anticholinergic drugs are limited by the ubiquitous nature of cholinergic pathways, and thus, by the inherent difficulty of effecting a specific pharmacologic intervention without inducing adverse effects. Nonetheless, medications with somewhat targeted cholinomimetic and anticholinergic actions are in widespread clinical use for their effects on the brain (especially cognition and behavior), neuromuscular junction, heart, eyes, lungs, and genitourinary and gastrointestinal tracts.
Other chapters that discuss applications of cholinergic pharmacology are Chapter 18, Pharmacology of Analgesia; Chapter 47, Integrative Inflammation Pharmacology: Peptic Ulcer Disease; and Chapter 48, Integrative Inflammation Pharmacology: Asthma.
 The year is 1744. Virginian settlers capture Chief Opechancanough, warrior chief of the Powhatans and uncle to Pocahontas. Opechancanough is considered a master tactician and has a reputation as a brutal warrior. One colonial correspondent portrays a different picture of the captured chief, however: “The excessive fatigues he encountered wrecked his constitution; his flesh became macerated; his sinews lost their tone and elasticity and his eyelids were so heavy that he could not see unless they were lifted up by his attendants . . . he was unable to walk; but his spirit, rising above the ruins of his body, directed [his followers] from the litter on which he was carried by his Indians.” During the confinement of Opechancanough to a prison in Jamestown, it is discovered that, after a period of inactivity, he is able to raise himself from the ground to a standing position.
The year is 1744. Virginian settlers capture Chief Opechancanough, warrior chief of the Powhatans and uncle to Pocahontas. Opechancanough is considered a master tactician and has a reputation as a brutal warrior. One colonial correspondent portrays a different picture of the captured chief, however: “The excessive fatigues he encountered wrecked his constitution; his flesh became macerated; his sinews lost their tone and elasticity and his eyelids were so heavy that he could not see unless they were lifted up by his attendants . . . he was unable to walk; but his spirit, rising above the ruins of his body, directed [his followers] from the litter on which he was carried by his Indians.” During the confinement of Opechancanough to a prison in Jamestown, it is discovered that, after a period of inactivity, he is able to raise himself from the ground to a standing position.
It is thought that the story of Opechancanough provides the earliest recorded description of myasthenia gravis, a neuromuscular disease resulting from the autoimmune production of antibodies directed against cholinergic receptors at the neuromuscular junction. In 1934, almost 2 centuries later, the English physician Mary Broadfoot Walker encounters several patients with similar symptoms of muscle weakness, which remind her of the symptoms of patients with tubocurare poisoning. Given her findings, Dr. Walker administers an antidote, physostigmine, to her immobile patients. The results are startling—within minutes, her patients are able to rise and walk across the room. Dr. Walker has discovered the first truly effective medication for myasthenia gravis. Despite the significance of her accomplishment, it is largely ridiculed by the scientific community because the treatment improves the symptoms of myasthenia gravis too rapidly and effectively to be believable. It is not until many years later that the scientific community comes to accept her findings.
Questions
1. Why do tubocurare poisoning and myasthenia gravis produce similar symptoms?
2. How does physostigmine improve the symptoms of myasthenia gravis? Why is it dangerous to administer physostigmine to every patient presenting with muscle weakness?
3. What are the therapeutic uses of anticholinergic drugs in other diseases such as Alzheimer’s dementia?
4. What are the advantages and disadvantages of using medications with anticholinergic effects in older individuals and individuals with cognitive impairments?
 BIOCHEMISTRY AND PHYSIOLOGY OF CHOLINERGIC NEUROTRANSMISSION
BIOCHEMISTRY AND PHYSIOLOGY OF CHOLINERGIC NEUROTRANSMISSION
Acetylcholine synthesis, storage, and release follow a similar set of steps in all cholinergic neurons. The specific effects of ACh at a particular cholinergic synapse are largely determined by the ACh receptor type at that synapse. Cholinergic receptors are divided into two broad classes. Muscarinic acetylcholine receptors (mAChR) are G protein-coupled receptors that are expressed at the terminal synapses of all parasympathetic postganglionic fibers and a few sympathetic postganglionic fibers, at autonomic ganglia, and in the CNS. Nicotinic acetylcholine receptors (nAChR) are ligand-gated ion channels that are concentrated postsynaptically at many excitatory autonomic synapses and presynaptically in the CNS. Acetylcholinesterase (AChE), the enzyme responsible for acetylcholine degradation, is also an important pharmacologic target. In this section, the biochemistry of each of these pharmacologic targets is described and the physiologic effects of acetylcholine at the neuromuscular junction, in the autonomic nervous system, in the CNS, and in the non-neuronal cholinergic system are discussed.
Acetylcholine is synthesized in a single step from choline and acetyl coenzyme A (acetyl CoA) by the enzyme choline acetyltransferase (ChAT):

In the CNS, choline used for the synthesis of acetylcholine arises from one of three sources. Approximately 35% to 50% of the choline generated by acetylcholinesterase in the synaptic cleft (see Fig. 10-1 and below) is transported back into the axon terminal, where it comprises about half of the choline used in ACh synthesis. Plasma-based stores of choline may also be transported to the brain as part of phosphatidylcholine (a phospholipid), which is then metabolized to free choline. (The incorporation of choline into phosphatidylcholine is essential, because choline itself cannot cross the blood–brain barrier.) Choline is also stored in phospholipids as phosphorylcholine, where it can be used when needed.
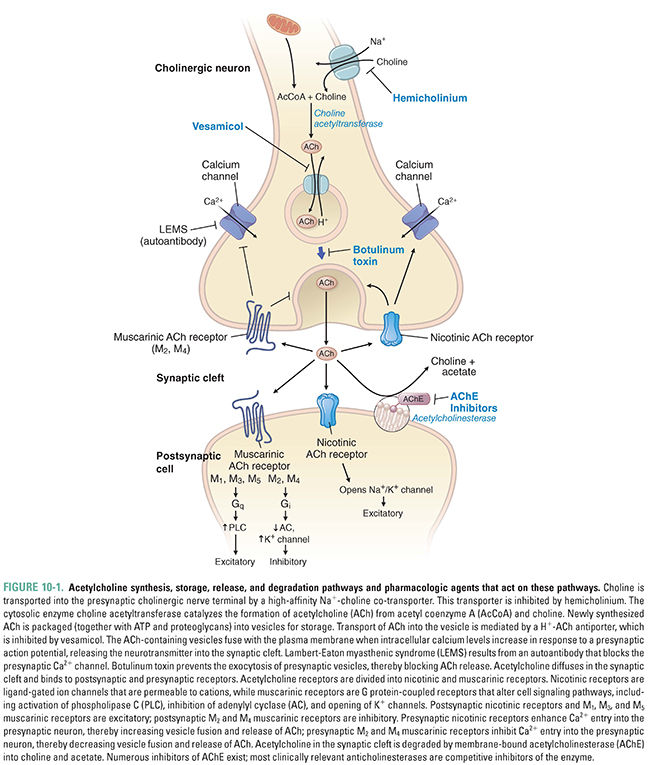
Acetyl CoA for the synthesis reaction is derived mainly from glycolysis and is ultimately produced by the enzyme pyruvate dehydrogenase. Although the synthesis of acetyl CoA occurs at the inner membrane of mitochondria, choline acetyltransferase is located in the cytoplasm. It is hypothesized that citrate serves as the carrier for acetyl CoA from the mitochondrion to the cytoplasm, where the citrate is freed by citrate lyase.
The choline acetyltransferase reaction is not the rate-limiting step in ACh synthesis. Rather, ACh synthesis is limited by the availability of the choline substrate, which depends on uptake of choline into the neuron. Two processes are responsible for choline transport. The first is low-affinity (Km = 10–100 μM) facilitated diffusion. This transport system is not saturable and is found in cells that synthesize choline-containing phospholipids, such as the corneal epithelium. Far more important is a sodium-dependent, high-affinity transport system (Km = 1–5 μM) found specifically in cholinergic nerve terminals. Because the high-affinity transporter is saturated at concentrations of choline >10 μM, it sets an upper limit on the supply of choline for ACh synthesis. As the rate-limiting component in ACh synthesis, this transporter is a target for several anticholinergic drugs (e.g., hemicholinium-3, see Fig. 10-1).
Storage and Release of Acetylcholine
After its synthesis in the cytoplasm, ACh is transported into synaptic vesicles for storage. An ATPase that pumps protons into the vesicle provides the energy necessary for this process. Transport of protons out of the vesicle (i.e., down the H+ concentration gradient) is coupled to uptake of ACh into the vesicle (i.e., against the ACh concentration gradient) via an ACh-H+ antiport channel. This antiporter is a target for some anticholinergic drugs, such as vesamicol, and its inhibition results in a deficit of ACh storage and subsequent release (Fig. 10-1). Cholinergic synaptic vesicles contain not only ACh but also ATP and heparan sulfate proteoglycans, both of which serve as counter-ions for ACh. By neutralizing the positive charge of ACh, these molecules disperse electrostatic forces that would otherwise prevent dense packing of ACh within the vesicle. (Released ATP also acts as a neurotransmitter, through purinergic receptors, to inhibit the release of ACh and norepinephrine from autonomic nerve endings.)
Release of ACh into the synaptic cleft occurs via fusion of the synaptic vesicle with the plasma membrane. This process depends on axon terminal depolarization and the opening of voltage-dependent calcium channels. The increase in intracellular Ca2+ facilitates the binding of synaptotagmin to the SNARE-complex proteins, which together mediate vesicle–membrane attachment and fusion. The result is that the contents of the vesicle are released as discrete “quanta” into the synaptic cleft. (See Chapter 8, Principles of Cellular Excitability and Electrochemical Transmission, for additional details on electrochemical transmission.)
Two stores of ACh have distinct roles during the process of ACh release. One store, known as the depot pool, consists of vesicles positioned near the plasma membrane of the axon terminal. Axonal depolarization causes these vesicles to release ACh rapidly. The reserve pool serves to refill the depot pool as it is being used. An adequate rate of reserve pool mobilization is required to sustain ACh release for an extended period of time. Of the two stores, the depot pool is replenished first by vesicles loaded with newly synthesized ACh; this process displaces some of the older depot pool vesicles into the reserve pool.
After ACh has been released into the synaptic cleft, it binds to one of two classes of receptors, usually on the membrane surface of the postsynaptic cell. Muscarinic acetylcholine receptors (mAChR) are seven-transmembrane-helix G protein-coupled receptors (GPCRs), and nicotinic acetylcholine receptors (nAChR) are ligand-gated ion channels. Although muscarinic receptors and nicotinic receptors are sensitive to the same neurotransmitter, these two classes of cholinergic receptors share little structural similarity.
Muscarinic cholinergic transmission occurs mainly at autonomic ganglia, at end organs innervated by the parasympathetic division of the autonomic nervous system, and in the CNS. As G protein-coupled receptors, muscarinic receptors transduce signals across the cell membrane and interact with GTP-binding proteins. Because the effects of muscarinic receptor activation occur through the actions of these G proteins, there is a latency of at least 100–250 ms associated with muscarinic responses to receptor activation. (In contrast, nicotinic receptor channels have latencies on the order of 5 ms.)
Activation of G proteins by agonist binding to muscarinic receptors may have several different effects on cells. These include inhibition of adenylyl cyclase (via Gi) and stimulation of phospholipase C (via Gq/11), both mediated by an α subunit of the G protein. (See Chapter 1, Drug–Receptor Interactions, for a discussion of these signaling mechanisms.) Muscarinic activation also modulates ion channels via the βγ subunit of a G protein. The predominant effect of such mAChR stimulation is to increase the opening of specific potassium channels (G protein-modulated inwardly rectifying K+ channels, or GIRKs), thereby hyperpolarizing the cell. The βγ subunit of the Gi protein binds to the channel and enhances its probability of being open.
Five distinct cDNAs for muscarinic receptors, denoted M1–M5, have been isolated and detected in human cells. These receptor types form two functionally distinct groups. M1, M3, and M5 are coupled to G proteins responsible for the stimulation of phospholipase C. M2 and M4, on the other hand, are coupled to G proteins responsible for adenylyl cyclase inhibition and K+ channel activation. The receptors of each functional group can be distinguished based on their responses to pharmacologic antagonists (Table 10-1). Generally, M1 is expressed in cortical neurons and autonomic ganglia, M2 in cardiac muscle, and M3 in smooth muscle and glandular tissue. Because stimulation of M1, M3, and M5 receptors facilitates excitation of the cell, while stimulation of M2 and M4 receptors suppresses cellular excitability, there is a predictable correlation between the receptor subtype and the effect of ACh on the cell. The various muscarinic receptor subtypes account for much of the diversity in cellular responses to mAChR agonists.
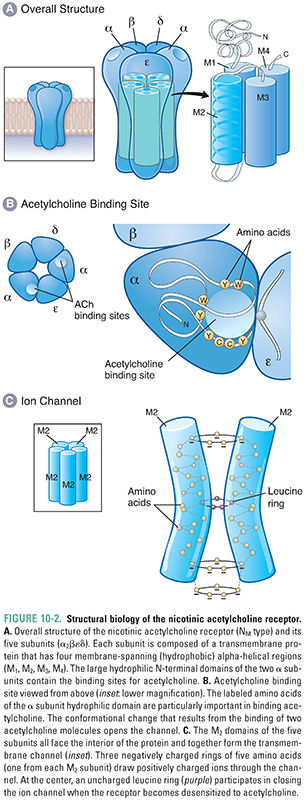
Nicotinic acetylcholine receptors (nAChRs) mediate nicotinic cholinergic transmission via a process known as direct ligand-gated conductance (Fig. 10-2). The binding of two ACh molecules to one nAChR elicits a conformational change in the receptor that creates a monovalent cation-selective pore through the cell membrane. Open channels of the activated nAChR are equally permeable to K+ and Na+ ions. (Since the resting membrane potential is close to the Nernst potential for K+ and far below the Nernst potential for Na+, the predominant ion passing through the open nACR is Na+.) A relatively small permeability to Ca2+ ions also results in important elevations of intracellular [Ca2+]. Therefore, when open, these channels produce a net inward current that depolarizes the postsynaptic cell. Stimulation of multiple nAChRs may depolarize the cell sufficiently to generate action potentials and to open voltage-dependent calcium channels. The latter action, and the direct entry of Ca2+ through the nAChR pore, can lead to activation of several intracellular signaling pathways.
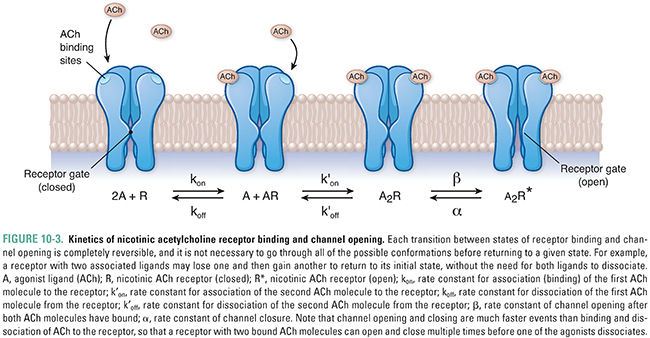
Because ACh dissociates rapidly from active-state receptor molecules and acetylcholinesterase rapidly degrades free (unbound) ACh in the synaptic cleft (see below), the depolarization mediated by nAChRs is brief (<10 ms). Although the simultaneous binding of two ACh molecules is required for channel opening, it is not necessary for both molecules to dissociate from the receptor in order for the channel to open again; the binding of a second ACh molecule to a receptor that still has one ACh bound may, once again, result in channel opening. The kinetics of nAChR binding and channel opening are detailed in Figure 10-3.
Structurally, the nicotinic acetylcholine receptor comprises five subunits, each of which has a mass of approximately 40 kilodaltons (Fig. 10-2A). Several types of nAChR subunits have been identified; these are designated α, β, γ, δ, and ε. All of these subunits share 35–50% homology with one another. Each receptor at the NMJ is composed of two α subunits, one β and one δ subunit, and either one γ or one ε subunit. (The α2βεδ form dominates at the neuromuscular junction in mature skeletal muscle, while the α2βγδ form is expressed in embryonic muscle.) Agonist molecules bind at a hydrophobic pocket that is formed between each α subunit and the adjacent, complementary subunit—this is the structural basis for the binding of two ACh molecules to each receptor. The conformational change in the α subunits induced by the binding of ACh initiates the overall changes in the pore that permit ion flow through the receptor (i.e., that open the channel).
Besides simply opening and closing in response to ACh binding, nicotinic receptors also modulate their responses to various concentration profiles of ACh. The receptors react differently to discrete, brief pulses of ACh than to neurotransmitter that is present continuously. As noted above, under normal conditions, a closed, resting-state channel responds to dual ACh binding by opening transiently, and the low affinity of the receptor for ACh causes rapid dissociation of ACh from the receptor and return of the receptor to its resting conformation. In contrast, continuous exposure of the receptor to ACh causes it to undergo a change to a “desensitized” conformation in which the channel is locked closed. The desensitized state is also characterized by a greatly increased affinity of the receptor for ACh, so that ACh remains bound to the receptor for a relatively long period of time. This prolonged binding of ACh to the desensitized conformation delays the conversion of the receptor to its resting state and hence prolongs the time during which the receptor is incapable of being activated by agonist.
Nicotinic cholinergic receptors at autonomic ganglia and in the central nervous system (termed N2 or NN) are similar to receptors at the NMJ (N1 or NM), with the exception that the subunits in NN receptors are composed solely of α and β subunits. To complicate matters, however, nine different α subunit types (α2–α10) and three β subunit types (β2–β4) have been detected in neuronal tissues. (α1 and β1 refer to the distinct subunit types found at the NMJ.) This diversity of α and β subunit combinations is responsible for the variable responses of CNS and autonomic nAChRs to pharmacologic agents. Presynaptic nAChRs in the CNS modulate the release both of ACh itself and of other excitatory and inhibitory neurotransmitters. This effect may involve prolonged elevations of [Ca2+] in the presynaptic nerve terminals, which lead to inactivation of neuronal calcium channels.
In order for acetylcholine to be useful for rapid, repeated neurotransmission, there must be a mechanism to limit its duration of action. Degradation of ACh is essential not only to prevent unwanted activation of neighboring neurons or muscle cells but also to ensure proper timing of signaling at the postsynaptic cell. A single receptor molecule is typically capable of distinguishing between two sequential presynaptic release events because degradation of ACh in the synaptic cleft occurs faster than the time course of nAChR activation.
Enzymes collectively known as cholinesterases are responsible for degrading acetylcholine. The two types of cholinesterase, AChE and butyrylcholinesterase (BuChE, also known as pseudocholinesterase or nonspecific cholinesterase), are distributed widely throughout the body. AChE is indispensable for the degradation of ACh and is capable of hydrolyzing about 4 × 105 molecules of ACh per enzyme molecule per minute; its turnover time of 150 μs makes it one of the most efficient hydrolytic enzymes known. AChE is concentrated on the postsynaptic membrane, and the choline that it frees there is efficiently transported back into the presynaptic terminal. BuChE has a secondary role in ACh degradation; the enzyme can hydrolyze ACh but at rates much slower than that of AChE. Evidence suggests that BuChE may be involved in early neural development as a co-regulator of ACh and may also be involved in the pathogenesis of Alzheimer’s disease. Because of its central importance to cholinergic transmission, a class of drugs known as acetylcholinesterase inhibitors has been designed to target AChE.
Physiologic Effects of Cholinergic Transmission
Acetylcholine is the principal neurotransmitter at the neuromuscular junction (Fig. 10-4). ACh is released by α motor neurons and it binds to nicotinic receptors in the muscle cell membrane, resulting in motor end-plate depolarization. The extent of depolarization depends on the quantity of ACh released into the synaptic cleft. Release of ACh is quantal in nature; that is, ACh is released in discrete quantities by the presynaptic motor neuron. Each quantum of ACh corresponds to the contents of a single synaptic vesicle and elicits a small depolarization in the motor end-plate termed a miniature end-plate potential (MEPP). Under resting conditions, sporadic MEPPs are detected at the motor end-plate, corresponding to a low baseline level of unstimulated ACh release that arises from spontaneous synaptic vesicle fusion with the motor axon’s presynaptic membrane. In contrast, the arrival of an action potential at the motor axon terminal causes many more vesicles (up to thousands) to fuse with the neuronal membrane and release their ACh. At the motor end-plate, the result is a relatively large depolarization termed the end-plate potential (EPP) (Fig. 10-5). The magnitude of an EPP is more than sufficient to trigger an action potential that propagates from the end-plate throughout the muscle fiber and, hence, produces a single contraction or “twitch.”
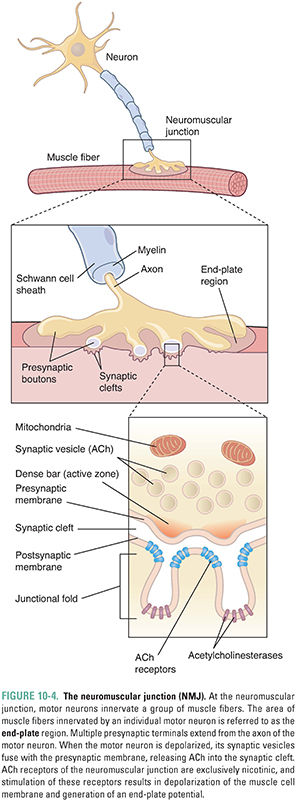
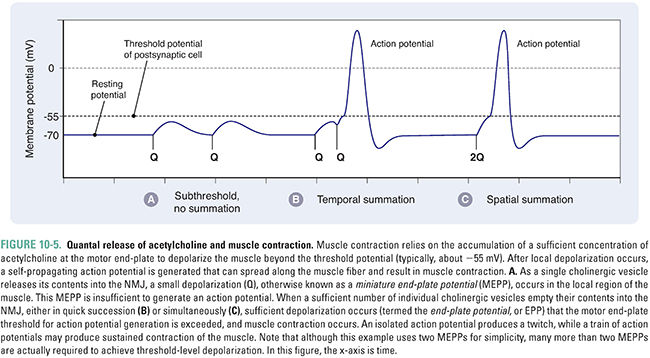
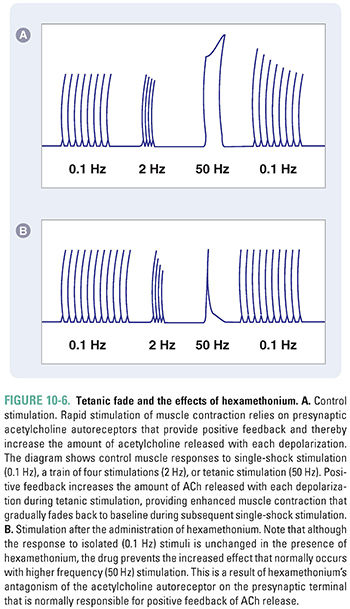
Acetylcholine not only triggers muscle contraction as its primary effect at the NMJ, but also modulates its own action at this site. Presynaptic cholinergic receptors, located on the axon terminal of the motor neuron, respond to ACh binding by facilitating the mobilization of synaptic vesicles from the reserve pool to the depot pool. This positive feedback loop, in which the release of ACh stimulates additional ACh release, is necessary to ensure sufficient ACh release when the nerve is stimulated with high frequency (~100 Hz). Despite this mechanism, the ACh output per nerve impulse wanes rapidly during prolonged high-frequency stimulation. Fortunately, because an excess of ACh is released and an excess of ACh receptors is present, there is a large safety margin. Only when 50% or more of the postsynaptic receptors are desensitized is a decline in muscle tension observed during tetanic stimulation (a phenomenon known as tetanic fade). Importantly, selective blockade of the modulatory presynaptic cholinergic receptors by antagonists such as hexamethonium prevents facilitation and causes rapid tetanic fade to occur under otherwise normal conditions (Fig. 10-6).
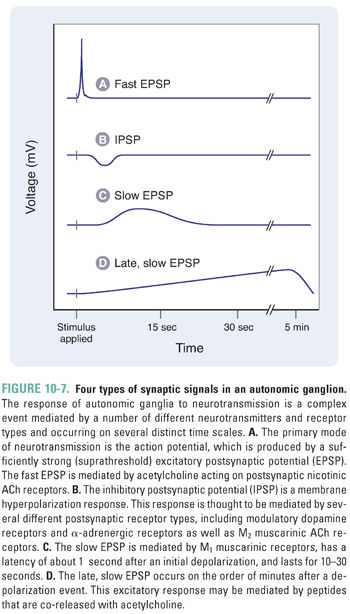
Autonomic activity can be classified as either tonic activity, which accounts for end organ stimulation at rest, or phasic activity, which triggers an elevated response to changing conditions. Neurotransmission through autonomic ganglia is complicated because several distinct receptor types contribute to the complex changes observed in postganglionic neurons. The generalized postsynaptic response to presynaptic impulses can be separated into four distinct components (Fig. 10-7). The primary event in the postsynaptic ganglionic response is a rapid depolarization mediated by nicotinic ACh receptors in the cell membrane of postganglionic neurons. The mechanism is similar to that in the NMJ, in that an inward current elicits a near-immediate excitatory postsynaptic potential (EPSP) of 10–50 ms in duration. Typically, the amplitude of such an EPSP is only a few millivolts, and many such events must sum for the postsynaptic cell membrane to reach the threshold for firing an action potential (Fig. 10-7A). The three remaining events of ganglionic transmission modulate this primary signal and are known as the slow EPSP, the IPSP (inhibitory postsynaptic potential), and the late, slow EPSP. The slow EPSP occurs after a latency of 1 second and is mediated by M1 muscarinic ACh receptors. The duration of this effect is 10–30 seconds (Fig. 10-7C). The IPSP is largely a product of catecholamine (i.e., dopamine and norepinephrine) stimulation of dopaminergic and α-adrenergic receptors (see Chapter 11, Adrenergic Pharmacology), although some IPSPs in a few ganglia are mediated by M2 muscarinic receptors. The latency and duration of the IPSPs generally vary between those of the fast and slow EPSPs. The late, slow EPSP is mediated by a decrease in potassium conductance induced by stimulation of receptors for peptide transmitters (i.e., angiotensin, substance P, and luteinizing hormone-releasing hormone). Lasting for several minutes, the late, slow EPSP is thought to have a role in the long-term regulation of postsynaptic neuron sensitivity to repetitive depolarization.
One pharmacologic consequence of such a complex pattern of depolarization in autonomic ganglia is that drugs selective for the IPSP, slow EPSP, and late, slow EPSP are generally not capable of eliminating ganglionic transmission. Instead, such agents alter only the efficiency of transmission. For example, methacholine, a muscarinic receptor agonist, has modulatory effects on autonomic ganglia that resemble the stimulation of slow EPSPs (see below). Blockade of excitatory transmission through autonomic ganglia relies on inhibition of the nAChRs that mediate fast EPSPs.
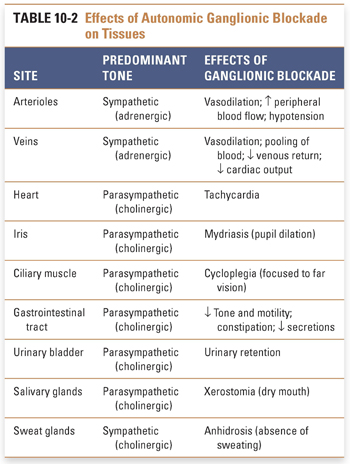
The overall effect of ganglionic blockade is complex and depends on the relative predominance of sympathetic and parasympathetic tone at the various end organs (Table 10-2). For example, the heart is influenced at rest primarily by the parasympathetic system, whose tonic effect is a slowing of the heart rate. Thus, blockade of autonomic ganglia that innervate the heart by moderate to high doses of the antimuscarinic agent atropine results in blockade of vagal slowing of the sinoatrial node and hence in relative tachycardia. (It should be noted that in low doses, the central parasympathetic stimulating effects of atropine predominate, initially resulting in bradycardia prior to its peripheral vagolytic action.) Blood vessels, in contrast, are innervated only by the sympathetic system. Because the normal effect of sympathetic stimulation is to cause vasoconstriction, ganglionic blockade results in vasodilation. It is important to realize, however, that the responses described above ignore the presence of muscarinic ACh receptors at many of the end organs. When stimulated directly by cholinergic agents, such receptors often mediate a response that overrides the response produced by ganglionic blockade. In general, the expected net cardiovascular effects of muscarinic blockade produced by clinical doses of atropine in a healthy adult with a normal hemodynamic state are mild tachycardia, with or without flushing of the skin, and no profound effect on blood pressure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


