54
Cancer Chemotherapy
CASE STUDY
A 55-year-old man presents with increasing fatigue, 15-pound weight loss, and a microcytic anemia. Colonoscopy identifies a mass in the ascending colon, and biopsy specimens reveal well-differentiated colorectal cancer (CRC). He undergoes surgical resection and is found to have high-risk stage III CRC with five positive lymph nodes. After surgery, he feels entirely well with no symptoms. Of note, he has no other comorbid illnesses. What is this patient’s prognosis? Should he receive adjuvant chemotherapy? The patient receives a combination of 5-fluorouracil (5-FU), leucovorin, and oxaliplatin as adjuvant therapy. One week after receiving the first cycle of therapy, he experiences significant toxicity in the form of myelosuppression, diarrhea, and altered mental status. What is the most likely explanation for this increased toxicity? Is there any role for genetic testing to determine the etiology of this level of toxicity?
In 2014, approximately 1.6 million new cancer cases will be diagnosed in the USA, and nearly 580,000 individuals will die from this disease. Cancer is the second most common cause of death in the United States, accounting for 1 in 4 deaths. It is a disease characterized by a defect in the normal control mechanisms that govern cell survival, proliferation, and differentiation. Cells that have undergone neoplastic transformation usually express cell surface antigens that may be of normal fetal type, and they may display other signs of apparent immaturity, and may exhibit qualitative or quantitative chromosomal abnormalities, including various translocations and the appearance of amplified gene sequences. It is now well established that a small subpopulation of cells, referred to as tumor stem cells, reside within a tumor mass. They retain the ability to undergo repeated cycles of proliferation as well as to migrate to distant sites in the body to colonize various organs in the process called metastasis. Such tumor stem cells thus can express clonogenic (colony-forming) capability, and they are characterized by chromosome abnormalities reflecting their genetic instability, which leads to progressive selection of subclones that can survive more readily in the multicellular environment of the host. This genetic instability also allows them to become resistant to chemotherapy and radiotherapy. The invasive and metastatic processes as well as a series of metabolic abnormalities associated with the cancer result in tumor-related symptoms and eventual death of the patient unless the neoplasm can be eradicated with treatment.
CAUSES OF CANCER
The incidence, geographic distribution, and behavior of specific types of cancer are related to multiple factors, including sex, age, race, genetic predisposition, and exposure to environmental carcinogens. Of these factors, environmental exposure is probably most important. Exposure to ionizing radiation has been well documented as a significant risk factor for a number of cancers, including acute leukemias, thyroid cancer, breast cancer, lung cancer, soft tissue sarcoma, and basal cell and squamous cell skin cancers. Chemical carcinogens (particularly those in tobacco smoke) as well as azo dyes, aflatoxins, asbestos, benzene, and radon have all been well documented as leading to a wide range of human cancers.
Several viruses have been implicated in the etiology of various human cancers. For example, hepatitis B (HBV) and hepatitis C (HCV) are associated with the development of hepatocellular cancer; HIV is associated with Hodgkin’s and non-Hodgkin’s lymphomas; human papillomavirus (HPV) is associated with cervical cancer, anal and penile cancers, and oropharyngeal head and neck cancer; and Ebstein-Barr virus, also known as human herpesvirus 4 (HHV-4), is associated with nasopharyngeal cancer, Burkitt’s lymphoma, and Hodgkin’s lymphoma. Expression of virus-induced neoplasia may also depend on additional host and environmental factors that modulate the transformation process. Cellular genes are known that are homologous to the transforming genes of the retroviruses, a family of RNA viruses, and induce oncogenic transformation. These mammalian cellular genes, known as oncogenes, have been shown to code for specific growth factors and their corresponding receptors. These genes may be amplified (increased number of gene copies) or mutated, both of which can lead to constitutive overexpression in malignant cells. The bcl-2 family of genes represents a series of pro-survival genes that promotes survival by directly inhibiting apoptosis, a key pathway of programmed cell death.
ACRONYMS
Another class of genes, known as tumor suppressor genes, may be deleted or mutated, which gives rise to the neoplastic phenotype. The p53 gene is the best-established tumor suppressor gene identified to date, and the normal wild-type gene appears to play an important role in suppressing malignant transformation. Of note, p53 is mutated in up to 50% of all human solid tumors, including liver, breast, colon, lung, cervix, bladder, prostate, and skin.
CANCER TREATMENT MODALITIES
With present methods of treatment, when the tumor remains localized at the time of diagnosis, about one-third of patients are cured with local treatment strategies, such as surgery or radiotherapy. Earlier diagnosis might lead to increased cure rates with such local treatment. In the remaining cases, however, early micrometastasis is a characteristic feature, indicating that a systemic approach with chemotherapy is required for effective cancer management. In patients with locally advanced disease, chemotherapy is often combined with radiotherapy to allow for subsequent surgical resection to take place, and such a combined modality approach has led to improved clinical outcomes. At present, about 50% of patients who are initially diagnosed with cancer can be cured. In contrast, chemotherapy alone is able to cure less than 10% of all cancer patients when the tumor is diagnosed at an advanced stage.
Chemotherapy is presently used in three main clinical settings: (1) primary induction treatment for advanced disease or for cancers for which there are no other effective treatment approaches, (2) neoadjuvant treatment for patients who present with localized disease, for whom local forms of therapy such as surgery or radiation, or both, are inadequate by themselves, (3) adjuvant treatment to local methods of treatment, including surgery, radiation therapy, or both.
Primary chemotherapy refers to chemotherapy administered as the primary treatment in patients who present with advanced cancer for which no alternative treatment exists. This has been the main approach in treating patients with advanced metastatic disease, and in most cases, the goals of therapy are to relieve tumor-related symptoms, improve overall quality of life, and prolong time to tumor progression. Studies in a wide range of solid tumors have shown that chemotherapy in patients with advanced disease confers survival benefit when compared with supportive care, providing sound rationale for the early initiation of drug treatment. However, cancer chemotherapy can be curative in only a small subset of patients who present with advanced disease. In adults, these curable cancers include Hodgkin’s and non-Hodgkin’s lymphoma, acute myelogenous leukemia, germ cell cancer, and choriocarcinoma, while the curable childhood cancers include acute lymphoblastic leukemia, Burkitt’s lymphoma, Wilms’ tumor, and embryonal rhabdomyosarcoma.
Neoadjuvant chemotherapy refers to the use of chemotherapy in patients who present with localized cancer for which alternative local therapies, such as surgery, exist but which are less than completely effective. At present, neoadjuvant therapy is most often administered in the treatment of anal cancer, bladder cancer, breast cancer, esophageal cancer, laryngeal cancer, locally advanced non-small cell lung cancer (NSCLC), and osteogenic sarcoma. For some of these diseases, such as anal cancer, gastroesophageal cancer, laryngeal cancer, and NCSLC, optimal clinical benefit is derived when chemotherapy is administered with radiation therapy either concurrently or sequentially. The goal of the neoadjuvant approach is to reduce the size of the primary tumor so that surgical resection can then be made easier. In addition, in some cases such as with rectal cancer and laryngeal cancer, the administration of combined modality therapy prior to surgery can result in sparing of vital organs such as the rectum or larynx. In most cases, additional chemotherapy is given after surgery has been performed.
One of the most important roles for cancer chemotherapy is as an adjuvant to local treatment modalities such as surgery, and this has been termed adjuvant chemotherapy. In this setting, chemotherapy is administered after surgery has been performed, and the goal of chemotherapy is to reduce the incidence of both local and systemic recurrence and to improve the overall survival of patients. In general, chemotherapy regimens with clinical activity against advanced disease may have curative potential following surgical resection of the primary tumor, provided the appropriate dose and schedule are administered. Adjuvant chemotherapy is effective in prolonging both disease-free survival (DFS) and overall survival (OS) in patients with breast cancer, colon cancer, gastric cancer, NCSLC, Wilms’ tumor, anaplastic astrocytoma, and osteogenic sarcoma. Patients with primary malignant melanoma at high risk of local recurrence or systemic metastases derive clinical benefit from adjuvant treatment with the biologic agent α-interferon, although this treatment must be given for 1 year’s duration for maximal clinical efficacy. Finally, the antihormonal agents tamoxifen, anastrozole, and letrozole are effective in the adjuvant therapy of postmenopausal women with early-stage breast cancer whose breast tumors express the estrogen receptor (see Chapter 40 for additional details). However, because these agents are cytostatic rather than cytocidal, they must be administered on a long-term basis, with the standard recommendation being 5 years’ duration.
ROLE OF CELL CYCLE KINETICS & ANTI-CANCER EFFECT
The key principles of cell cycle kinetics were initially developed using the murine L1210 leukemia as the experimental model system (Figure 54–1). However, drug treatment of human cancers requires a clear understanding of the differences between the characteristics of this rodent leukemia and of human cancers, as well as an understanding of the differences in growth rates of normal target tissues between mice and humans. For example, L1210 is a rapidly growing leukemia with a high percentage of cells synthesizing DNA, as measured by the uptake of tritiated thymidine (the labeling index). Because L1210 leukemia has a growth fraction of 100% (ie, all its cells are actively progressing through the cell cycle), its life cycle is consistent and predictable. Based on the murine L1210 model, the cytotoxic effects of anti-cancer drugs follow log cell-kill kinetics. As such, a given agent would be predicted to kill a constant fraction of cells as opposed to a constant number.
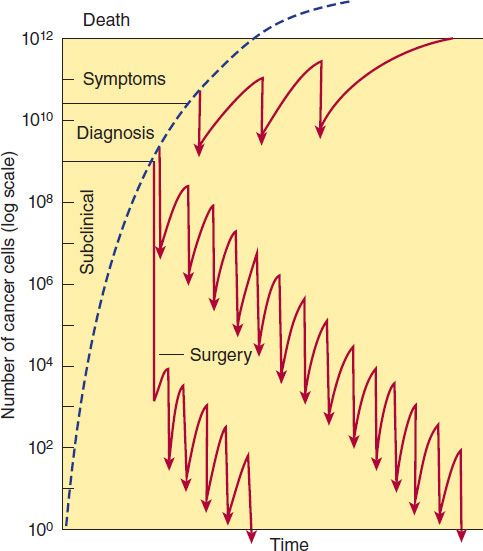
FIGURE 54–1 Log-kill hypothesis—relationship of tumor cell number to time of diagnosis, symptoms, treatment, and survival. Three alternative approaches to drug treatment are shown for comparison with the course of tumor growth when no treatment is given (dashed line). In the protocol diagrammed at top, treatment (indicated by the arrows) is given infrequently, and the result is manifested as prolongation of survival but with recurrence of symptoms between courses of treatment and eventual death of the patient. The combination chemotherapy treatment diagrammed in the middle section is begun earlier and is more intensive. Tumor cell kill exceeds regrowth, drug resistance does not develop, and “cure” results. In this example, treatment has been continued long after all clinical evidence of cancer has disappeared (1–3 years). This approach has been established as effective in the treatment of childhood acute leukemia, testicular cancers, and Hodgkin’s lymphoma. In the treatment diagrammed near the bottom of the graph, early surgery has been employed to remove the primary tumor and intensive adjuvant chemotherapy has been administered long enough (up to 1 year) to eradicate the remaining tumor cells that comprise the occult micrometastases.
Thus, if a particular dose of an individual drug leads to a 3-log kill of cancer cells and reduces the tumor burden from 1010 to 107 cells, the same dose used at a tumor burden of 105 cells reduces the tumor mass to 102 cells. Cell kill is, therefore, proportional, regardless of tumor burden. The cardinal rule of chemotherapy—the invariable inverse relation between cell number and curability—was established with this model, and this relationship is applicable to other hematologic malignancies.
Although growth of murine leukemias simulates exponential cell kinetics, mathematical modeling data suggest that most human solid tumors do not grow in such an exponential manner. Taken together, the experimental data in human solid cancers support a Gompertzian model of tumor growth and regression. The critical distinction between Gompertzian and exponential growth is that the growth fraction of the tumor is not constant with Gompertzian kinetics but instead decreases exponentially with time (exponential growth is matched by exponential retardation of growth, due to blood supply limitations and other factors). The growth fraction peaks when the tumor is approximately one-third its maximum size. Under the Gompertzian model, when a patient with advanced cancer is treated, the tumor mass is larger, its growth fraction is low, and the fraction of cells killed is, therefore, small. An important feature of Gompertzian growth is that response to chemotherapy in drug-sensitive tumors depends, in large measure, on where the tumor is in its particular growth curve.
Information on cell and population kinetics of cancer cells explains, in part, the limited effectiveness of most available anticancer drugs. A schematic summary of cell cycle kinetics is presented in Figure 54–2. This information is relevant to the mode of action, indications, and scheduling of cell cycle-specific (CCS) and cell cycle-nonspecific (CCNS) drugs. Agents falling into these two major classes are summarized in Table 54–1.
TABLE 54–1 Cell cycle effects of major classes of anti-cancer drugs.
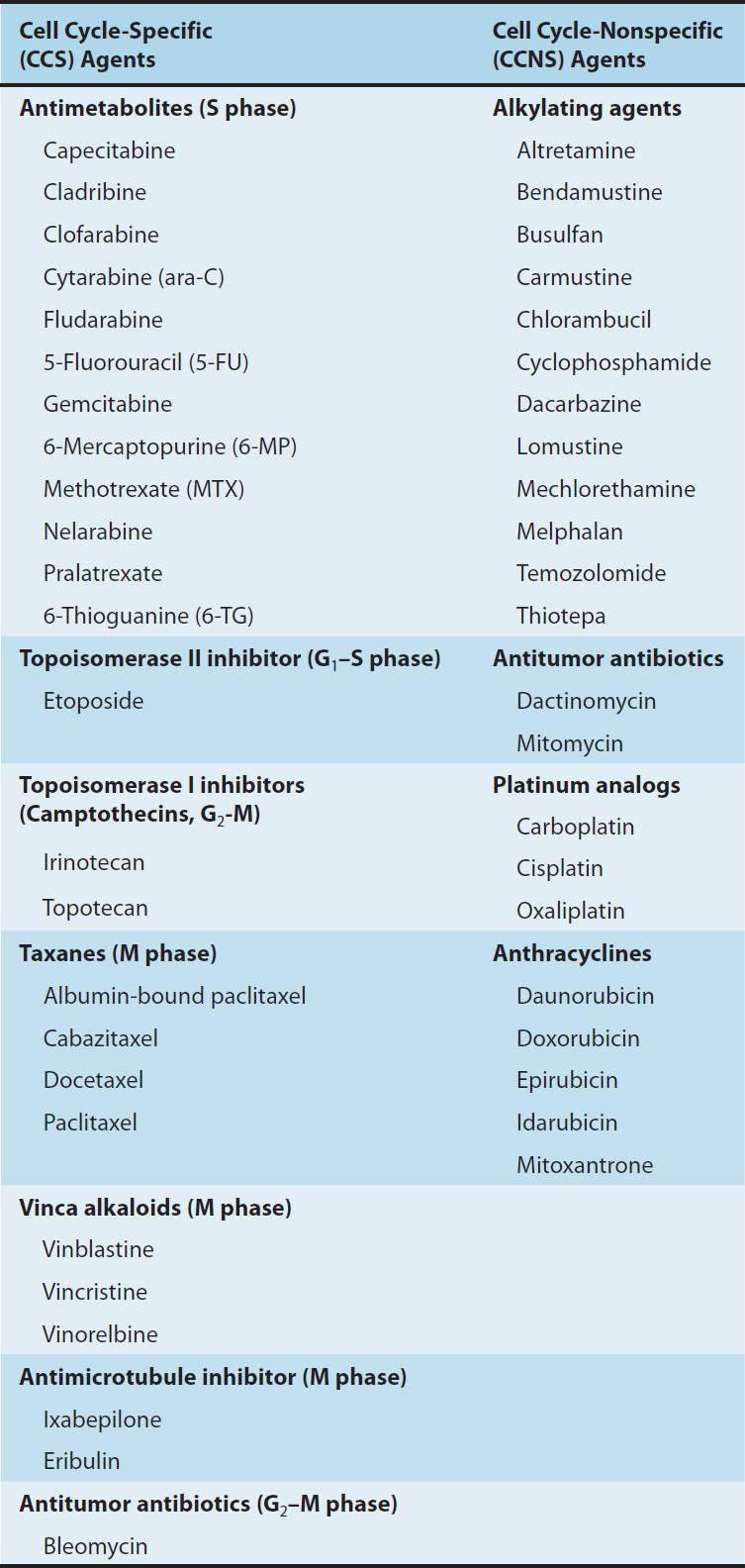
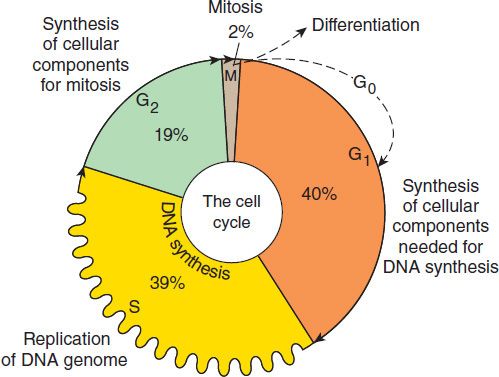
FIGURE 54–2 Cell cycle and cancer. A conceptual depiction of the cell cycle phases that all cells—normal and neoplastic—must traverse before and during cell division. The percentages given represent the approximate percentage of time spent in each phase by a typical malignant cell; the duration of G1, however, can vary markedly. Many of the effective anti-cancer drugs exert their action on cells traversing the cell cycle and are called cell cycle-specific (CCS) drugs (see Table 54–1). A second group of agents called cell cycle-nonspecific (CCNS) drugs can sterilize tumor cells whether they are cycling or resting in the G0 compartment. CCNS drugs can kill both G0 and cycling cells (although cycling cells are more sensitive).
The Role of Drug Combinations
With rare exceptions (eg, choriocarcinoma and Burkitt’s lymphoma), single drugs at clinically tolerable doses have been unable to cure cancer. In the 1960s and early 1970s, drug combination regimens were developed based on the known biochemical actions of available anti-cancer drugs rather than on their clinical efficacy. Such regimens were, however, largely ineffective. The era of effective combination chemotherapy began when a number of active drugs from different classes became available for use in combination in the treatment of the acute leukemias and lymphomas. Following this initial success with hematologic malignancies, combination chemotherapy was extended to the treatment of solid tumors.
The use of combination chemotherapy is important for several reasons. First, it provides maximal cell kill within the range of toxicity tolerated by the host for each drug as long as dosing is not compromised. Second, it provides a broader range of interaction between drugs and tumor cells with different genetic abnormalities in a heterogeneous tumor population. Finally, it may prevent or slow the subsequent development of cellular drug resistance. The same principles apply to the therapy of chronic infections, such as HIV and tuberculosis.
Certain principles have guided the selection of drugs in the most effective drug combinations, and they provide a paradigm for the development of new drug therapeutic programs.
1. Efficacy: Only drugs known to be somewhat effective when used alone against a given tumor should be selected for use in combination. If available, drugs that produce complete remission in some fraction of patients are preferred to those that produce only partial responses.
2. Toxicity: When several drugs of a given class are available and are equally effective, a drug should be selected on the basis of toxicity that does not overlap with the toxicity of other drugs in the combination. Although such selection leads to a wider range of adverse effects, it minimizes the risk of a lethal effect caused by multiple insults to the same organ system by different drugs and allows dose intensity to be maximized.
3. Optimum scheduling: Drugs should be used in their optimal dose and schedule, and drug combinations should be given at consistent intervals. Because long intervals between cycles negatively affect dose intensity, the treatment-free interval between cycles should be the shortest time necessary for recovery of the most sensitive normal target tissue, which is usually the bone marrow.
4. Mechanism of interaction: There should be a clear understanding of the biochemical, molecular, and pharmacokinetic mechanisms of interaction between the individual drugs in a given combination, to allow for maximal effect. Omission of a drug from a combination may allow overgrowth by a tumor clone sensitive to that drug alone and resistant to other drugs in the combination.
5. Avoidance of arbitrary dose changes: An arbitrary reduction in the dose of an effective drug in order to add other less effective drugs may reduce the dose of the most effective agent below the threshold of effectiveness and destroy the ability of the combination to cure disease in a given patient.
Dosage Factors
Dose intensity is one of the main factors limiting the ability of chemotherapy or radiation therapy to achieve cure. As described in Chapter 2, the dose-response curve in biologic systems is usually sigmoidal in shape, with a threshold, a linear phase, and a plateau phase. For chemotherapy, therapeutic selectivity is dependent on the difference between the dose-response curves of normal and tumor tissues. In experimental animal models, the dose-response curve is usually steep in the linear phase, and a reduction in dose when the tumor is in the linear phase of the dose-response curve almost always results in a loss in the capacity to cure the tumor effectively before a reduction in the antitumor activity is observed. Although complete remissions continue to be observed with dose reduction down to as low as 20% of the optimal dose, residual tumor cells may not be entirely eliminated, thereby allowing for eventual relapse. Because anti-cancer drugs are associated with toxicity, it is often appealing for clinicians to avoid acute toxicity by simply reducing the dose or by increasing the time interval between each cycle of treatment. However, such empiric modifications in dose represent a major cause of treatment failure in patients with drug-sensitive tumors.
A positive relationship between dose intensity and clinical efficacy has been documented in several solid tumors, including advanced ovarian, breast, lung, and colon cancers, as well as in hematologic malignancies, such as the lymphomas. At present, there are three main approaches to dose-intense delivery of chemotherapy. The first approach, dose escalation, involves increasing the doses of the respective anti-cancer agents. The second strategy is administration of anti-cancer agents in a dose-intense manner by reducing the interval between treatment cycles, while the third approach involves sequential scheduling of either single agents or of combination regimens. Each of these strategies is presently being applied to a wide range of solid cancers, including breast, colorectal, and NSCLC, and in general, such dose-intense regimens have significantly improved clinical outcomes.
DRUG RESISTANCE
A fundamental problem in cancer chemotherapy is the development of cellular drug resistance. Primary, or inherent resistance refers to drug resistance in the absence of prior exposure to available standard agents. The presence of inherent drug resistance was first proposed by Goldie and Coleman in the early 1980s and was thought to result from the genomic instability associated with the development of most cancers. For example, mutations in the p53 tumor suppressor gene occur in up to 50% of all human tumors. Preclinical and clinical studies have shown that loss of p53 function leads to resistance to radiation therapy as well as resistance to a wide range of anti-cancer agents. Defects in the mismatch repair enzyme family, which are tightly linked to the development of familial and sporadic colorectal cancer, are associated with resistance to several unrelated anti-cancer agents, including the fluoropyrimidines, the thiopurines, and cisplatin/carboplatin. In contrast to primary resistance, acquired resistance develops in response to exposure to a given anti-cancer agent. Experimentally, drug resistance can be highly specific to a single drug and is usually based on a specific change in the genetic machinery of a given tumor cell with amplification or increased expression of one or more genes. In other instances, a multidrug-resistant phenotype occurs, associated with increased expression of the MDR1 gene, which encodes a cell surface transporter glycoprotein (P-glycoprotein, see Chapter 5). This form of drug resistance leads to enhanced drug efflux and reduced intracellular accumulation of a broad range of structurally unrelated anti-cancer agents, including the anthracyclines, vinca alkaloids, taxanes, camptothecins, epipodophyllotoxins, and even small molecule inhibitors, such as imatinib.
 BASIC PHARMACOLOGY OF CANCER CHEMOTHERAPEUTIC DRUGS
BASIC PHARMACOLOGY OF CANCER CHEMOTHERAPEUTIC DRUGS
ALKYLATING AGENTS
The major clinically useful alkylating agents (Figure 54–3) have a structure containing a bis(chloroethyl)amine, ethyleneimine, or nitrosourea moiety, and they are classified in several different groups. Among the bis(chloroethyl)amines, cyclophosphamide, mechlorethamine, melphalan, and chlorambucil are the most useful. Ifosfamide is closely related to cyclophosphamide but has a somewhat different spectrum of activity and toxicity. Thiotepa and busulfan are used to treat breast and ovarian cancer, and chronic myeloid leukemia, respectively. The major nitrosoureas are carmustine (BCNU) and lomustine (CCNU).
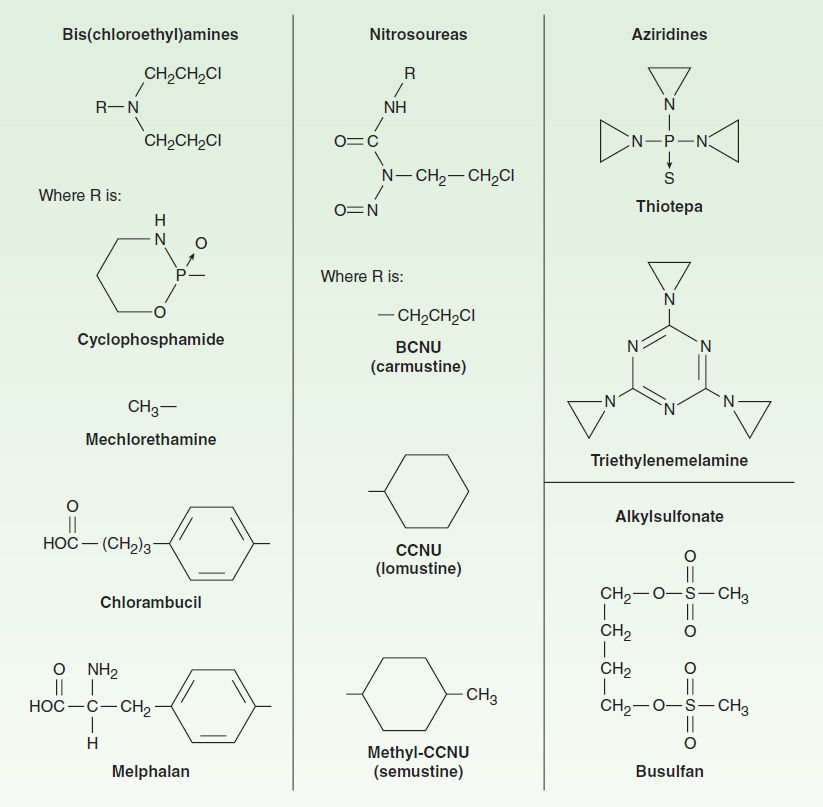
FIGURE 54–3 Structures of major classes of alkylating agents.
Mechanism of Action
As a class, the alkylating agents exert their cytotoxic effects via transfer of their alkyl groups to various cellular constituents. Alkylations of DNA within the nucleus probably represent the major interactions that lead to cell death. However, these drugs react chemically with sulfhydryl, amino, hydroxyl, carboxyl, and phosphate groups of other cellular nucleophiles as well. The general mechanism of action of these drugs involves intramolecular cyclization to form an ethyleneimonium ion that may directly or through formation of a carbonium ion transfer an alkyl group to a cellular constituent (Figure 54–4). In addition to alkylation, a secondary mechanism that occurs with nitrosoureas involves carbamoylation of lysine residues of proteins through formation of isocyanates.
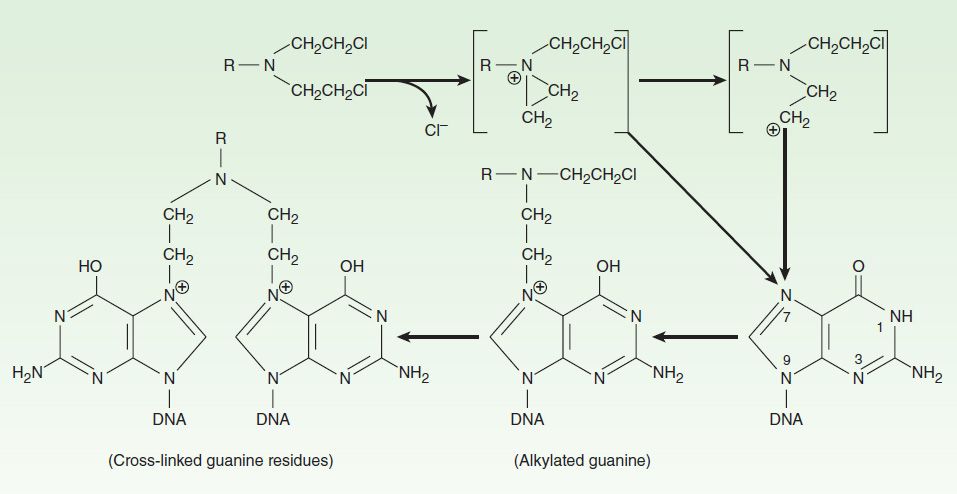
FIGURE 54–4 Mechanism of alkylation of DNA guanine. A bis(chloroethyl)amine forms an ethyleneimonium ion that reacts with a base such as N7 of guanine in DNA, producing an alkylated purine. Alkylation of a second guanine residue, through the illustrated mechanism, results in cross-linking of DNA strands.
The major site of alkylation within DNA is the N7 position of guanine; however, other bases are also alkylated albeit to lesser degrees, including N1 and N3 of adenine, N3 of cytosine, and O6 of guanine, as well as phosphate atoms and proteins associated with DNA. These interactions can occur on a single strand or on both strands of DNA through cross-linking, as most major alkylating agents are bifunctional, with two reactive groups. Alkylation of guanine can result in miscoding through abnormal base pairing with thymine or in depurination by excision of guanine residues. The latter effect leads to DNA strand breakage through scission of the sugar-phosphate backbone of DNA. Cross-linking of DNA appears to be of major importance to the cytotoxic action of alkylating agents, and replicating cells are most susceptible to these drugs. Thus, although alkylating agents are not cell cycle-specific, cells are most susceptible to alkylation in late G1 and S phases of the cell cycle.
Resistance
The mechanism of acquired resistance to alkylating agents may involve increased capability to repair DNA lesions through increased expression and activity of DNA repair enzymes, decreased transport of the alkylating drug into the cell, and increased expression or activity of glutathione and glutathione-associated proteins, which are needed to conjugate the alkylating agent, or increased glutathione S-transferase activity, which catalyzes the conjugation.
Adverse Effects
The adverse effects usually associated with alkylating agents are generally dose-related and occur primarily in rapidly growing tissues such as bone marrow, gastrointestinal tract, and reproductive system. Nausea and vomiting can be a serious issue with a number of these agents. In addition, they are potent vesicants and can damage tissues at the site of administration as well as produce systemic toxicity. As a class, alkylating agents are carcinogenic in nature, and there is an increased risk of secondary malignancies, especially acute myelogenous leukemia.
Cyclophosphamide is one of the most widely used alkylating agents. One of the potential advantages of this compound relates to its high oral bioavailability. As a result, it can be administered via the oral and intravenous routes with equal clinical efficacy. It is inactive in its parent form, and must be activated to cytotoxic forms by liver microsomal enzymes (Figure 54–5). The cytochrome P450 mixed-function oxidase system converts cyclophosphamide to 4-hydroxycyclophosphamide, which is in equilibrium with aldophosphamide. These active metabolites are delivered to both tumor and normal tissue, where nonenzymatic cleavage of aldophosphamide to the cytotoxic forms—phosphoramide mustard and acrolein—occurs. The liver appears to be protected through the enzymatic formation of the inactive metabolites 4-ketocyclophosphamide and carboxyphosphamide.
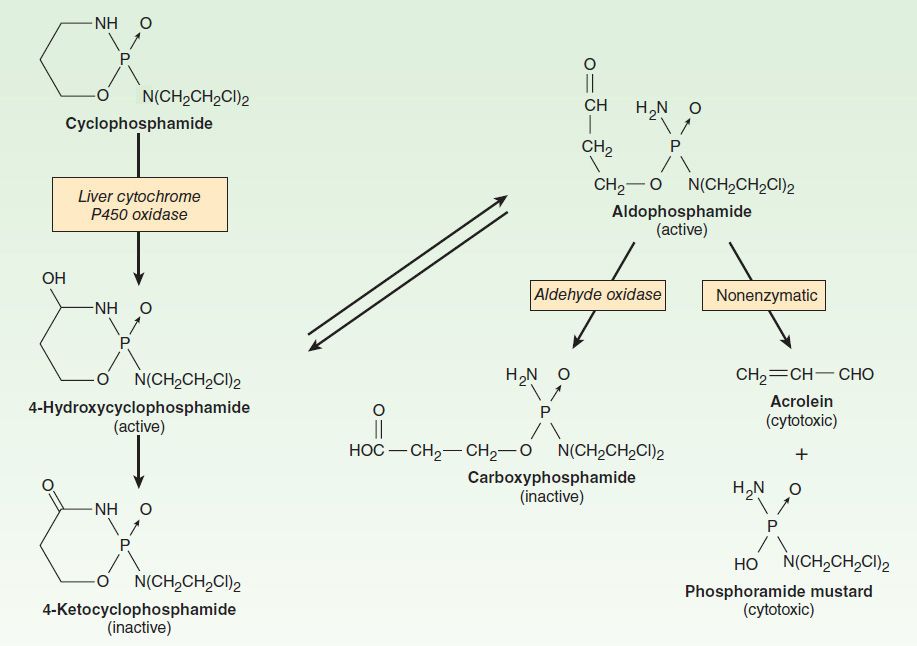
FIGURE 54–5 Cyclophosphamide metabolism.
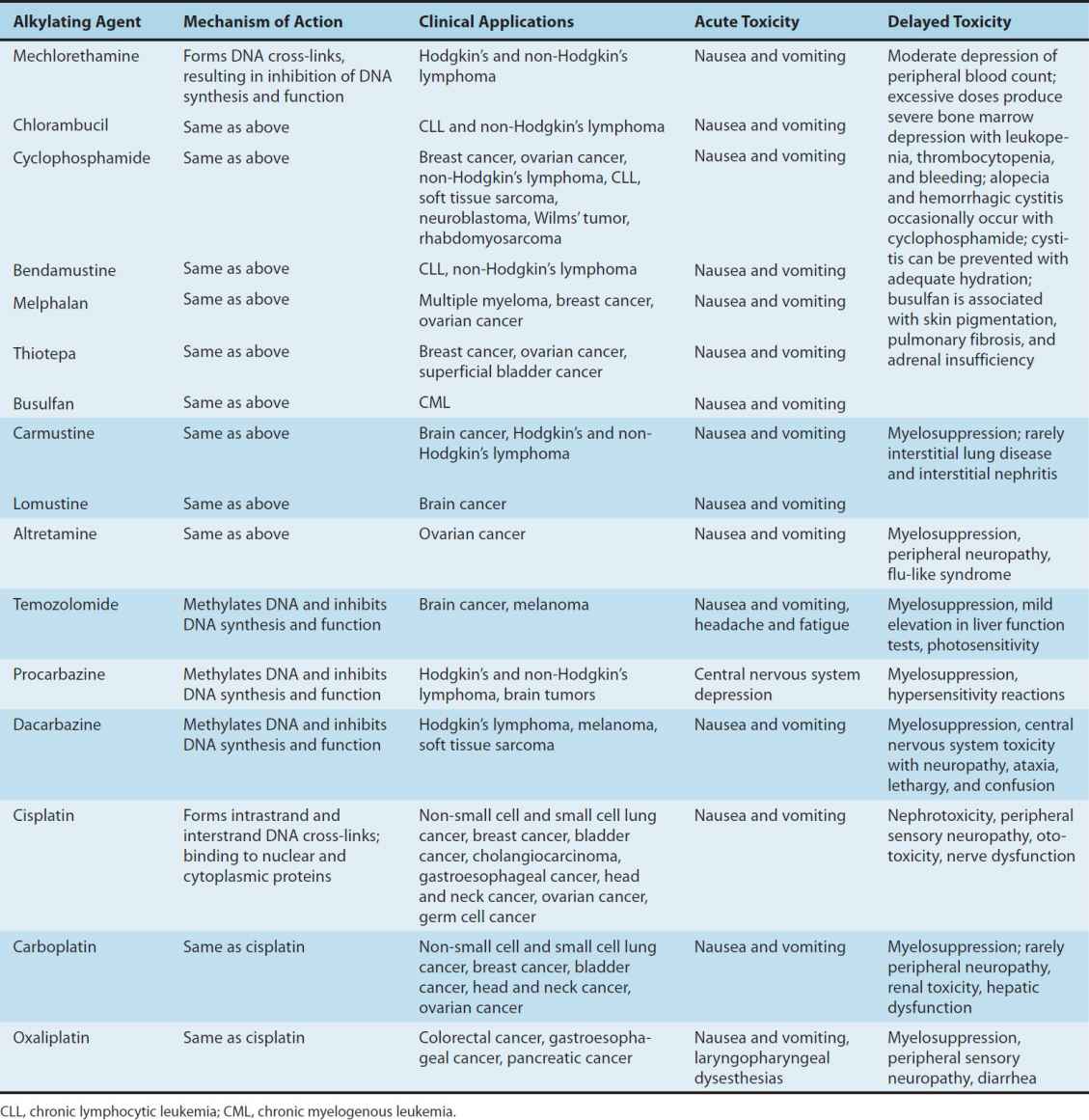
The major toxicities of the individual alkylating agents are outlined in Table 54–2 and discussed below.
TABLE 54–2 Alkylating agents and platinum analogs: Clinical activity and toxicities.
NITROSOUREAS
These drugs appear to be non-cross-resistant with other alkylating agents; all require biotransformation, which occurs by nonenzymatic decomposition, to metabolites with both alkylating and carbamoylating activities. The nitrosoureas are highly lipid-soluble and are able to readily cross the blood-brain barrier, making them effective in the treatment of brain tumors. Although the majority of alkylations by the nitrosoureas are on the N7 position of guanine in DNA, the critical alkylation responsible for cytotoxicity appears to be on the O6 position of guanine, which leads to G-C crosslinks in DNA. After oral administration of lomustine, peak plasma levels of metabolites appear within 1–4 hours; central nervous system concentrations reach 30–40% of the activity present in the plasma. Urinary excretion appears to be the major route of elimination from the body. One naturally occurring sugar-containing nitrosourea, streptozocin, is interesting because it has minimal bone marrow toxicity. This agent has activity in the treatment of insulin-secreting islet cell carcinoma of the pancreas.
NONCLASSIC ALKYLATING AGENTS
Several other compounds have mechanisms of action that involve DNA alkylation as their cytotoxic mechanism of action. These agents include procarbazine, dacarbazine, and bendamustine. Their clinical activities and toxicities are listed in Table 54–2.
Procarbazine
Procarbazine is an orally active methylhydrazine derivative, and in the clinical setting, it is used in combination regimens for Hodgkin’s and non-Hodgkin’s lymphoma as well as brain tumors.
The precise mechanism of action of procarbazine is uncertain; however, it inhibits DNA, RNA, and protein biosynthesis; prolongs interphase; and produces chromosome breaks. Oxidative metabolism of this drug by microsomal enzymes generates azoprocarbazine and H2O2, which may be responsible for DNA strand scission. A variety of other drug metabolites are formed that may be cytotoxic. One metabolite is a weak monoamine oxidase (MAO) inhibitor, and adverse events can occur when procarbazine is given with other MAO inhibitors as well as with sympathomimetic agents, tricyclic antidepressants, antihistamines, central nervous system depressants, antidiabetic agents, alcohol, and tyramine-containing foods.
There is an increased risk of secondary cancers in the form of acute leukemia, and its carcinogenic potential is thought to be higher than that of most other alkylating agents.
Dacarbazine
Dacarbazine is a synthetic compound that functions as an alkylating agent following metabolic activation in the liver by oxidative N-demethylation to the monomethyl derivative. This metabolite spontaneously decomposes to diazomethane, which generates a methyl carbonium ion that is believed to be the key cytotoxic species. Dacarbazine is administered parenterally and is used in the treatment of malignant melanoma, Hodgkin’s lymphoma, soft tissue sarcomas, and neuroblastoma. The main dose-limiting toxicity is myelosuppression, but nausea and vomiting can be severe in some cases. This agent is a potent vesicant, and care must be taken to avoid extravasation during drug administration.
Bendamustine
Bendamustine is a bifunctional alkylating agent consisting of a purine benzimidazole ring and a nitrogen mustard moiety. As with other alkylating agents, it forms cross-links with DNA resulting in single- and double-stranded breaks, leading to inhibition of DNA synthesis and function. This molecule also inhibits mitotic checkpoints and induces mitotic catastrophe, which leads to cell death. Of note, the cross-resistance between bendamustine and other alkylating agents is only partial, thereby providing a rationale for its clinical activity despite the development of resistance to other alkylating agents. This agent is approved for use in chronic lymphocytic leukemia, with activity also observed in Hodgkin’s and non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer. The main dose-limiting toxicities include myelosuppression and mild nausea and vomiting. Hypersensitivity infusion reactions, skin rash, and other skin reactions occur rarely.
PLATINUM ANALOGS
Three platinum analogs are currently used in clinical practice: cisplatin, carboplatin, and oxaliplatin. Cisplatin (cis-diamminedichloroplatinum [II]) is an inorganic metal complex that was initially discovered through a serendipitous observation that neutral platinum complexes inhibited division and filamentous growth of Escherichia coli. Several platinum analogs were subsequently synthesized. Although the precise mechanism of action of the platinum analogs is unclear, they are thought to exert their cytotoxic effects in the same manner as alkylating agents. As such, they kill tumor cells in all stages of the cell cycle and bind DNA through the formation of intrastrand and interstrand cross-links, thereby leading to inhibition of DNA synthesis and function. The primary binding site is the N7 position of guanine, but covalent interaction with the N3 position of adenine and O6 position of cytosine can also occur. In addition to targeting DNA, the platinum analogs have been shown to bind to both cytoplasmic and nuclear proteins, which may also contribute to their cytotoxic and antitumor effects. The platinum complexes appear to synergize with certain other anti-cancer drugs, including alkylating agents, fluoropyrimidines, and taxanes. The major toxicities of the individual platinum analogs are outlined in Table 54–2.
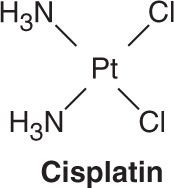
Cisplatin has major antitumor activity in a broad range of solid tumors, including non-small cell and small cell lung cancer, esophageal and gastric cancer, cholangiocarcinoma, head and neck cancer, and genitourinary cancers, particularly testicular, ovarian, and bladder cancer. When used in combination regimens, cisplatin-based therapy has led to the cure of nonseminomatous testicular cancer. Cisplatin and the other platinum analogs are extensively cleared by the kidneys and excreted in the urine. As a result, dose modification is required in patients with renal dysfunction.
Carboplatin is a second-generation platinum analog whose mechanisms of cytotoxic action, mechanisms of resistance, and clinical pharmacology are identical to those described for cisplatin. As with cisplatin, carboplatin has broad-spectrum activity against a wide range of solid tumors. However, in contrast to cisplatin, it exhibits significantly less renal toxicity and gastrointestinal toxicity. Its main dose-limiting toxicity is myelosuppression. It has therefore been widely used in transplant regimens to treat refractory hematologic malignancies. Moreover, since vigorous intravenous hydration is not required for carboplatin therapy, carboplatin is viewed as an easier agent to administer to patients, and as such, it has replaced cisplatin in various combination chemotherapy regimens.
Oxaliplatin is a third-generation diaminocyclohexane platinum analog. Its mechanism of action and clinical pharmacology are identical to those of cisplatin and carboplatin. However, tumors that are resistant to cisplatin or carboplatin on the basis of mismatch repair defects are not cross-resistant to oxaliplatin, and this finding may explain the activity of this platinum compound in colorectal cancer. Oxaliplatin was initially approved for use as second-line therapy in combination with the fluoropyrimidine 5-fluorouracil (5-FU) and leucovorin, termed the FOLFOX regimen, for metastatic colorectal cancer. There are various iterations of the FOLFOX regimen, which have now become the most widely used combination regimens in the first-line treatment of advanced colorectal cancer. In addition, this regimen is widely used in the adjuvant therapy of stage III colon cancer and high-risk stage II colon cancer. Clinical activity has also been observed in other gastrointestinal cancers, such as pancreatic, gastroesophageal, and hepatocellular cancer. Neurotoxicity is the main dose-limiting toxicity, and it is manifested by a peripheral sensory neuropathy. There are two forms of neurotoxicity, an acute form that is often triggered and worsened by exposure to cold, and a chronic form that is dose-dependent. Although this chronic form is dependent on the cumulative dose of drug administered, it tends to be reversible, in contrast to cisplatin-induced neurotoxicity.
ANTIMETABOLITES
The development of drugs with actions on intermediary metabolism of proliferating cells has been important both conceptually and clinically. While biochemical properties unique to all cancer cells have yet to be discovered, there are a number of quantitative differences in metabolism between cancer cells and normal cells that render cancer cells more sensitive to the antimetabolites. Many of these agents have been rationally designed and synthesized based on knowledge of critical cellular processes involved in DNA biosynthesis. The individual antimetabolites and their respective clinical spectrum and toxicities are presented in Table 54–3 and are discussed below.
TABLE 54–3 Antimetabolites: Clinical activity and toxicities.
ANTIFOLATES
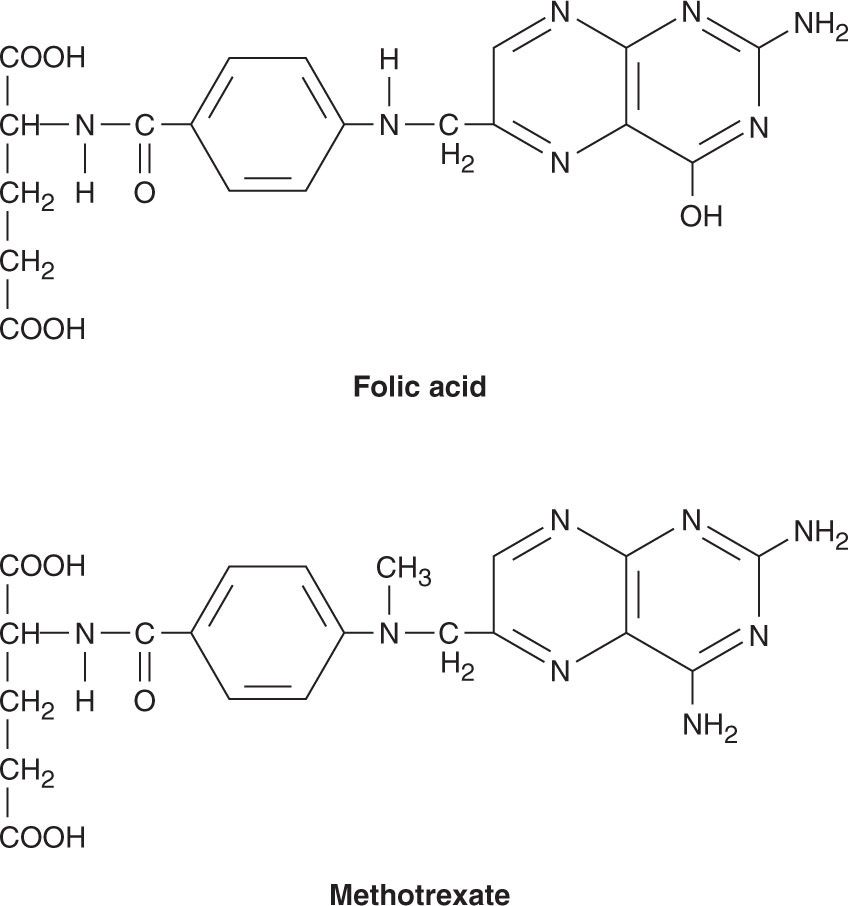
Methotrexate
Methotrexate (MTX) is a folic acid analog that binds with high affinity to the active catalytic site of dihydrofolate reductase (DHFR). This results in inhibition of the synthesis of tetrahydrofolate (THF), the key one-carbon carrier for enzymatic processes involved in de novo synthesis of thymidylate, purine nucleotides, and the amino acids serine and methionine. Inhibition of these metabolic processes thereby interferes with the formation of DNA, RNA, and key cellular proteins (see Figure 33–3). Intracellular formation of polyglutamate metabolites, with the addition of up to 5–7 glutamate residues, is critically important for the therapeutic action of MTX, and this process is catalyzed by the enzyme folylpolyglutamate synthase (FPGS). MTX polyglutamates are selectively retained within cancer cells, and they display increased inhibitory effects on enzymes involved in de novo purine nucleotide and thymidylate biosynthesis, making them important determinants of MTX’s cytotoxic action.
Several resistance mechanisms to MTX have been identified, and they include (1) decreased drug transport via the reduced folate carrier or folate receptor protein, (2) decreased formation of cytotoxic MTX polyglutamates, (3) increased levels of the target enzyme DHFR through gene amplification and other genetic mechanisms, and (4) altered DHFR protein with reduced affinity for MTX. Recent studies have suggested that decreased accumulation of drug through activation of the multidrug resistance transporter P170 glycoprotein may also result in drug resistance.
MTX is administered by the intravenous, intrathecal, or oral route. However, oral bioavailability is saturable and erratic at doses greater than 25 mg/m2. Renal excretion is the main route of elimination and is mediated by glomerular filtration and tubular secretion. As a result, dose modification is required in the setting of renal dysfunction. Care must also be taken when MTX is used in the presence of drugs such as aspirin, nonsteroidal anti-inflammatory agents, penicillin, and cephalosporins, as these agents inhibit the renal excretion of MTX. The biologic effects of MTX can be reversed by administration of the reduced folate leucovorin (5-formyltetrahydrofolate) or by L-leucovorin, which is the active enantiomer. Leucovorin rescue is used in conjunction with high-dose MTX therapy to rescue normal cells from undue toxicity, and it has also been used in cases of accidental drug overdose. The main adverse effects are listed in Table 54–3.
Pemetrexed
Pemetrexed is a pyrrolopyrimidine antifolate analog with activity in the S phase of the cell cycle. As in the case of MTX, it is transported into the cell via the reduced folate carrier and requires activation by FPGS to yield higher polyglutamate forms. While this agent targets DHFR and enzymes involved in de novo purine nucleotide biosynthesis, its main mechanism of action is inhibition of thymidylate synthase (TS). At present, this antifolate is approved for use in combination with cisplatin in the treatment of mesothelioma, as a single agent in the second-line therapy of NSCLC, in combination with cisplatin for the first-line treatment of NSCLC, and most recently, as maintenance therapy in patients with NSCLC whose disease has not progressed after four cycles of platinum-based chemotherapy. As with MTX, pemetrexed is mainly excreted in the urine, and dose modification is required in patients with renal dysfunction. The main adverse effects include myelosuppression, skin rash, mucositis, diarrhea, fatigue, and hand-foot syndrome. Of note, vitamin supplementation with folic acid and vitamin B12
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


