Abbreviations used in text
Precursor B–cell lymphoblastic leukemia/lymphoma
Pre-B ALL/LBL
Mature B–cell neoplasms
Chronic lymphocytic leukemia/small lymphocytic lymphoma
CLL/SLL
B-cell prolymphocytic leukemia
PLL
Hairy cell leukemia
HCL
Lymphoplasmacytic lymphoma
LPL
Plasma cell myeloma/plasmacytoma
PCM
Splenic marginal zone lymphoma
SMZL
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue
MALToma
Nodal marginal zone lymphoma
NMZL
Follicular lymphoma
FL
Primary cutaneous follicle center lymphoma
PCFL
Mantle cell lymphoma
MCL
Diffuse large B-cell lymphoma
DLBCL
Lymphomatoid granulomatosis
LyG
Primary mediastinal (thymic) large B-cell lymphoma
PMLBCL
Intravascular large B-cell lymphoma
IBCL
ALK positive large B-cell lymphoma
ALK-DLBCL
Plasmablastic lymphoma
PBL
Large B-cell lymphoma arising in HHV-8-associated Castleman disease
HHV-8-DLBCL
Primary effusion lymphoma
PEL
Burkitt lymphoma
BL
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL
HGBCLU
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma
Immunodeficiency–associated B–cell lymphoproliferative disorders (LPD)
LPD associated with primary immune disorders
Lymphomas associated with HIV infection
Post-transplant LPD
PTLD
Other iatrogenic immunodeficiency-associated LPD
Molecular Basis of Disease
B-Cell Antigen Receptor Genes
AgRs are the primary effector molecules of the adaptive immune system and consist of multi-subunit glycoprotein molecules present on all T and B lymphocytes [2]. Each lymphocyte has a unique antigen receptor molecule on its membrane. B-cell AgRs are membrane-bound immunoglobulins (Ig), which include the heavy chain gene (IGH) at 14q32, the kappa light chain gene (IGK) at 2p11, and the lambda light chain gene (IGL) at 22q11. Each AgR gene in germline configuration consists of variable (V), joining (J), constant (C), and in the case of the IGH gene, diversity (D) regions. There are approximately 45 V, 23 D, and 6 J regions in the IGH gene. During B cell development, one IGH allele undergoes rearrangement, with deletion of large intervening DNA segments, followed by DNA repair at the site of recombination. This process is mediated by recombination activating genes (RAG1/2) which encode RAG1/2 proteins able to induce breaks in double stranded DNA. DNA damage response proteins then bring the separated AgR gene segments together and form a functional rearranged IGH gene [3]. AgR rearrangement occurs in a specific order: first, one IGH D segment is fused to one J segment, then this DJ segment is fused with a V segment to form a functional VDJ exon that encodes the variable antigen recognition site of the IGH protein. Following IGH rearrangement, the final product is a gene containing one V region, one J region, one D region, and multiple C regions, arranged in the following order on the chromosome: V-(D)-J-C (Fig. 42.1). Ig gene rearrangement is an error-prone process and many attempted rearrangements result in nonfunctional AgR genes. If the first IGH rearrangement in a cell fails, the second IGH allele is rearranged. A single B cell may therefore have two IGH rearrangements, one nonfunctional and one functional. The IGK alleles rearrange in a similar fashion, but only after the successful rearrangement of one of the IGH alleles. The IGL alleles rearrange in most cases only if rearrangements of the IGK genes fail and both alleles are deleted [4]. Normal B cells that fail to produce a functional IGH or light chain rearrangement are usually eliminated through apoptosis.
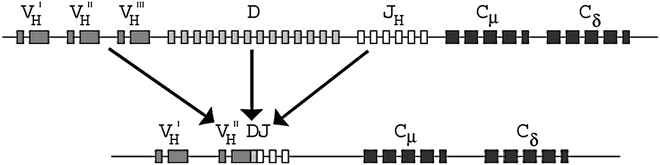
Figure 42.1
Configuration and rearrangement of the IGH gene on chromosome 14q32. DJ rearrangement occurs initially, followed by VD rearrangement. The rearrangements occur in a random fashion across B cells, resulting in unique rearrangement in each normal B cell. The rearranged IGH gene contains one VH segment (plus all the upstream VH segments), one DH segment, one JH segment (plus all the downstream JH segments), and the CH segments, in that order on the chromosome
After successful rearrangement, the Ig genes in mature B cells often undergo additional changes, including somatic hypermutation. In response to antigen exposure, somatic hypermutation occurs in the IGH V region (IGVH) of an already rearranged IGH gene in germinal center-derived B cells to enhance antigen affinity of the AgR. Somatic hypermutation occurs through point mutations, small insertions, small deletions, or some combination of these. The enzyme activation-induced cytidine deaminase (AID) mediates the process of both class-switching and somatic hypermutation. AID expression is upregulated in germinal centers in response to antigen stimuli [3, 4]. The term “postgerminal center” is used to refer to B cells that have been exposed to antigen in the germinal center and undergone additional somatic mutation of IGVH. Errors in class switch recombination, somatic hypermutation, and AID expression contribute to the pathogenesis of many B cell lymphomas [5].
Structurally and genetically unique AgR rearrangements occur in every B cell and are exploited as markers of cell lineage and clonality. B cells with these unique gene rearrangements may undergo limited clonal expansion as a part of a normal immune response, but uncontrolled clonal expansion occurs in BCL. In this setting, the unique AgR rearrangement can serve as a diagnostic marker of B-cell clonality and a marker for detection of MRD following therapy. Clonal rearrangements of IGH and IGK can be detected in essentially all malignancies of mature B cells, but many precursor B-cell malignancies will have only IGH rearrangements, since malignant transformation occurs before rearrangement of the IGK or IGL genes.
Recurrent Cytogenetic and Molecular Abnormalities in BCL and Plasma Cell Myeloma
Many, but not all BCL are associated with characteristic cytogenetic or molecular abnormalities that help to establish an accurate primary diagnosis, have specific prognostic significance, and/or serve as targets for MRD assessment. Most of the molecular abnormalities described below have been included under the National Comprehensive Cancer Network Guidelines (Version 3.2012) as “useful testing under certain circumstances for clarification of diagnosis.” Table 42.2 provides a summary of most of the recurrent cytogenetic abnormalities identified thus far in subtypes of BCL. This list illustrates the large number of potential targets for molecular testing, but also highlights the increasing challenge for clinical laboratories to provide relevant molecular information to clinicians. There are many additional newly discovered mutations in different BCL, either not yet proven to have clinical relevance and/or not yet part of routine testing algorithms in BCL; some of these are listed in Table 42.3 for the different BCL subtypes. The most clinically relevant BCL subtype-specific cytogenetic and molecular abnormalities, namely those with currently known diagnostic or prognostic significance, are discussed in more detail under the individual BCL categories below. General considerations that are applicable to most or all BCL subtypes are discussed later in the sections on diagnostic and prognostic indications for molecular testing in BCL.
Table 42.2
Nonrandom chromosomal abnormalities in B-cell malignancies
Lymphoma subtype | Nonrandom chromosomal alterations | Genes involved | |
|---|---|---|---|
CLL/SLL | Del 13q14 Trisomy 12 Del 11q22-23 Del 17p13 Del 6 3q27 | Unknown Unknown ATM TP53 | |
LPL | Del 6q Trisomy 4 | ||
MZL | t(11;18)(q21;q21) t(1;14)(p22;q32) t(14;18)(q32;q21) t(3;14)(q32) Trisomy 3 Trisomy 18 | API2–MALT1 BCL10–IGH IGH–MALT1 FOXP1 – IGH | |
FL | t(14;18)(q32;q21) | IGH–BCL2 | |
MCL | t(11;14)(q13;q32) | Cyclin D1 –IGH | |
DLBCL/BCLU | 3q27 rearrangement t(14;18)(q32;q21) 8q24 rearrangement | BCL6 IGH–BCL2 MYC | |
BL | t(8;14)(q24;q32) t(2;8)(p11;q24) t(8;22)(q24;q11) | MYC–IGH IGLK–MYC MYC–IGLL | |
PCM | t(11;14)(q13;q32) t(4;14)(p16;q32) t(14;16)(Q32;Q23) Monosomy 13 | Cyclin D1–IGH FGFR3+MMSET–IGH IGH–MUM1-IRF4 |
Table 42.3
Common gene mutations and deletions in B-cell lymphomas
MZL | CLL/SLL | MCL | FL | DLBCL–GCB | DLBCL–ABC |
|---|---|---|---|---|---|
TNFAIP3 – A20 | TP53 | TP53 | TP53 | TP53 | CD79B |
NOTCH2 | ATM | ATM | BCL2 | BCL2 | CARD11 |
NOTCH1 | NOTCH1 | EZH2 | EZH2 | MYD88 | |
SF3B1 | Cyclin D1 | CREBBP | CREBBP | TNFAIP3 – A20 | |
FBXW7 | p16 | EP300 | EP300 | INK4A – ARF | |
MYD88 | TNFAIP3 – A20 | MLL2 | MLL2 | BLIMP1 – PRDM1 | |
BIRC3 | MEF2B | MEF2B | BCL6 | ||
SGK1 | |||||
GNA13 | |||||
TNFRSF14 |
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Although morphologically and immunophenotypically similar, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is quite heterogeneous genetically, with different molecular abnormalities resulting in different natural histories and responses to therapy. New insights into the biology of CLL/SLL have improved the approach to treatment. Many of the relevant chromosomal abnormalities in CLL/SLL are not detected by karyotyping because the low proliferative capacity of most CLL/SLL limits the clinical utility of metaphase cytogenetics. The genetic abnormalities in CLL/SLL therefore usually are detected by interphase fluorescence in situ hybridization (FISH), which has become the clinical standard of care in CLL/SLL. Evaluation for these abnormalities is clinically indicated at diagnosis in most cases of CLL/SLL, because therapeutic decisions are affected by the molecular profile.
Adverse molecular prognostic factors in CLL/SLL include lack of IGVH hypermutation and the presence of TP53 or ATM gene mutations. Somatic hypermutation of IGVH is seen in approximately 50 % of CLL/SLL, and long-term progression-free and overall survival is better for patients with hypermutated IGVH. The explanation for this seems to be that unmutated CLL/SLL cells are genomically less stable than mutated CLL/SLL cells, are more likely to have acquired a TP53 deletion at relapse than those with mutated IGVH, and therefore are more likely to be resistant to salvage chemotherapy. Patients with intact TP53 gene function appear to have prolonged survival regardless of the extent of IGVH hypermutation [6]. Testing for IGVH hypermutation requires multiple PCR analyses and gene sequencing and is performed at only a few specialty laboratories and academic centers.
The TP53 gene deletion at 17p13.3 is identified by FISH testing in 7–15 % of CLL/SLL cases at diagnosis (illustrated in Fig. 42.2), but is detected by karyotype at a much lower rate. Other types of TP53 mutations also occur in many CLL/SLL including missense substitutions, frameshift mutations, and in frame deletions, and are highly correlated with 17p deletion; however, some TP53 mutations occur in the absence of deletion of 17p [7, 8]. TP53 maintains genome integrity by orchestrating the repair or elimination of cells with damaged DNA and contributes to the cytotoxicity of many anticancer agents. Both 17p deletion and inactivating mutations of TP53 render patients resistant to chemotherapy [9]. TP53 dysfunction is associated with an adverse clinical outcome in CLL/SLL patients, as well as in other BCL.
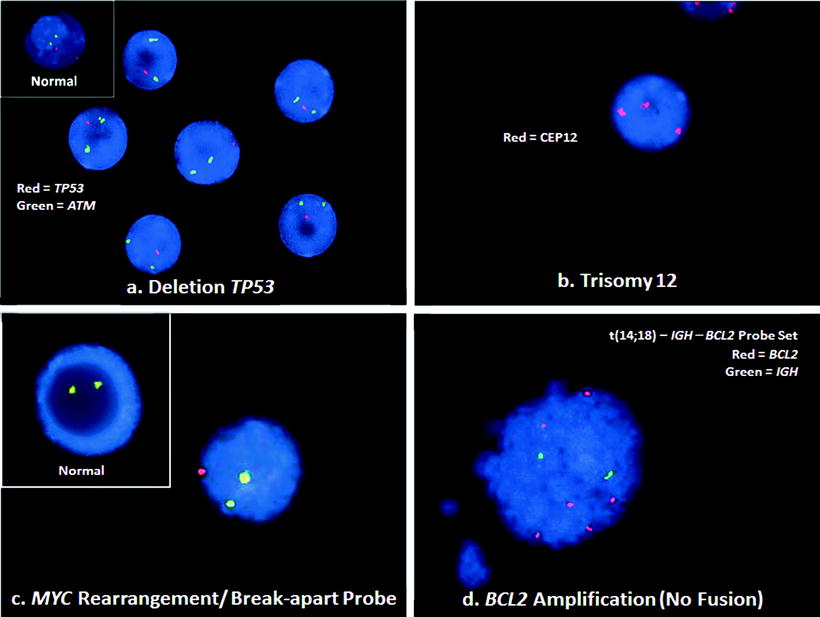
Figure 42.2
Illustrations of typical genetic abnormalities in BCL, detected by interphase FISH. (a) CLL/SLL lymphocytes with deletion of TP53. This case shows two green ATM (11q22.3) signals, but only one red TP53 (17p13.1) signal (RGG). A normal cell would have two red and two green signals (RRGG), as seen in the inset. (b) A centromeric probe (CEP12) for chromosome 12 demonstrates trisomy 12 with 3 red signals (RRR) in this cell from the blood of a CLL/SLL patient. A normal cell would have two red signals (RR). (c) A break-apart probe for the MYC gene shows a MYC rearrangement with a yellow fusion signal for the normal MYC and separate green and red signals for the rearranged MYC gene (RGY). The pattern in a normal cell with the break-apart probe would be two yellow signals (YY), shown in the inset. (d) BCL cell with BCL2 amplification. This FISH result does not show the t(14;18)– IGH – BCL2 fusion signals (RGYY) typical of the translocation. Rather, the pattern of six red signals for the BCL2 gene with only two signals for the IGH gene (6R2G) is consistent with BCL2 amplification. See Table 42.1 for abbreviations
Deletions in 11q22-23 are detected by FISH in 14–20 % of CLL/SLL cases and are associated with inactivation of the ataxia telangiectasia (ATM) gene. The ATM protein kinase is involved inTP53 regulation; therefore, ATM deletions produce TP53 dysfunction in CLL/SLL cells. ATM deletions most often are detected by FISH and are associated with a poor prognosis, although not as poor as that of patients with TP53 deletion/mutation. The addition of Rituximab monoclonal antibody therapy which targets CD20 on B cells to chemotherapy regimens appears to overcome the poor prognosis conferred by the 11q deletion [10, 11]. Mutations in ATM have been reported in approximately 10 % of CLL/SLL cases and only partially correlate with deletions in 11q. Approximately 18–30 % of cases with 11q deletion show concomitant ATM mutations. The significance of an ATM mutation in the absence of an 11q deletion is not well established.
As a single abnormality in CLL/SLL, 13q14 deletions convey the most favorable prognosis. CLL/SLL with 13q14 abnormalities usually has IGVH hypermutation. Detected by FISH, 13q14 deletions are seen in 40–55 % of CLL/SLL cases and are thought to affect one or more tumor suppressor genes. Frequent deletions and downregulation of microRNA genes miR15 and miR16 at 13q14 have been described in the majority of CLL/SLL cases with 13q14 deletions [12]. 13q14 deletions are sometimes seen in other BCL, but the specific gene(s) involved may not be the same.
Trisomy 12 can be detected by FISH or routine karyotyping and occurs in 16–20 % of CLL/SLL cases (FISH illustration in Fig. 42.2). Trisomy 12 usually is associated with unhypermutated IGVH and has been associated with atypical CLL/SLL morphology. Patients with trisomy 12 have an intermediate prognosis, with a median survival exceeded only by patients with single 13q14 abnormalities [10, 11].
Deletions of chromosome 6q are present in approximately 7 % of CLL/SLL cases and can be detected by FISH. The majority are identified at 6q21-23, with a smaller percentage documented at 6q25-27. Although limited, initial studies suggest that this class of cytogenetic abnormality is associated with an intermediate risk profile. Incorporation of FISH probes for 6q in clinical practice may improve our understanding of this molecular abnormality in risk stratification of patients with CLL/SLL.
In addition to the genetic abnormalities described above, exome and genome sequencing studies have identified additional recurrent gene mutations in CLL/SLL (Table 42.3) including those in SF3B1, NOTCH1, and others [13]. Frameshift mutations in the proline-glutamic acid-serine-threonine (PEST) degradation domain of NOTCH1 are associated with unhypermutated IGVH, trisomy 12, and a poor prognosis. Missense mutations in SF3B1, a gene involved in RNA splicing, are relatively common in CLL/SLL (approximately 10 %) and whereas the data are preliminary, there is evidence that these mutations also confer a worse clinical outcome [13–15].
B-Cell Prolymphocytic Leukemia
B-cell prolymphocytic leukemia (B-PLL) is an aggressive mature B-cell leukemia characterized by splenomegaly with minimal to no lymphadenopathy and prolymphocytes comprising > 55 % of circulating lymphoid cells. Complex karyotypes are common, as are TP53 mutations, 13q14 deletions, and to a lesser extent, trisomy 12. Cases with t(11;14)(q13;q32) are generally excluded from this category and considered leukemic variants of mantle cell lymphoma (MCL). The MYC gene can be overexpressed in B-PLL [16] and, although rare, the t(8;14)(q24;q32) translocation that involves MYC and is characteristic of Burkitt lymphoma (BL) has been reported in B-PLL [17, 18]. The significance of MYC abnormalities in B-PLL is not entirely clear and additional study is needed to elucidate the genetic events underlying the pathophysiology of this disease.
Hairy Cell Leukemia
Hairy cell leukemia (HCL) is an indolent neoplasm of mature B cells characterized by circumferential “hairlike” projections of the B-cell cytoplasmic membrane, splenomegaly, diffuse marrow involvement, and peripheral cytopenias. The BRAF V600E point mutation is present in nearly all cases of HCL and has redefined a disease which previously had no known recurrent molecular abnormalities. The BRAF V600E mutation is rare in other mature BCL [19]. The apparent specificity of this mutation for HCL makes this a disease-defining mutation with clear diagnostic utility, in addition to possible implications for MRD detection and treatment.
Lymphoplasmacytic Lymphoma
In the 2008 WHO classification, lymphoplasmacytic lymphoma (LPL) is defined as a B-cell neoplasm composed of a mixture of small lymphocytes, plasmacytoid lymphocytes, and plasma cells, that does not fulfill the criteria for any of the other malignancies of small B lymphocytes, i.e., a diagnosis of exclusion [1]. LPL includes Waldenström macroglobulinemia (WM). There are several common cytogenetic abnormalities in LPL, but none are specific for this lymphoma subtype. These abnormalities include 6q deletion, which is seen in approximately 50 % of cases presenting in the bone marrow (BM), less often in cases presenting in lymph nodes, and may be associated with a worse prognosis [20]. Trisomy 4 is present in 20 % of WM [21]. The t(9;14) translocation previously reported in LPL has been reassessed and confirmed not to be associated with LPL [22]. Gene expression profiling studies using flow cytometric sorted cells have shown that WM B cells cluster with CLL/SLL B cells, whereas WM plasma cells segregate with plasma cell myeloma cells [23]. One of the upregulated genes in WM B cells is interleukin-6, a molecule that may be a therapeutic target in LPL. WM is sometimes associated with clonal expansions of cytotoxic T cells in the blood, which can confound diagnosis. These T-cell clones can be eliminated by chemotherapy for LPL [24]. A mutation in MYD88 is present in almost all patients with WM. This mutation in MYD88 at 3p22.2 results in an amino acid change (L265P), and triggers NFKB signaling. The MYD88 L265P mutation is absent or only occasionally expressed in other subtypes of non-IgM LPL, as well as plasma cell myelomas and marginal zone lymphomas [25].
Plasma Cell Myeloma/Plasmacytoma
Plasma cell myeloma (PCM) and related plasma cell neoplasms have two major subgroups by cytogenetic and molecular characteristics with some overlap. The first group is hyperdiploid PCM, which comprises 55–60 % of cases and has trisomy or tetrasomy, predominantly of odd numbered chromosomes. The second group has chromosomal translocations that involve the IGH gene on 14q32, with a variety of partner genes. These translocations occur in approximately 50 % of PCM and the different translocations are sometimes associated with specific clinical features and outcome. Most cannot be identified by karyotyping, but are readily identified by FISH studies utilizing molecular probes for 14q32 and its translocation partners. Translocations involving the IGH locus in PCM are believed to occur at the time of isotype class switching, in contrast to other B-cell malignancies.
The t(11;14)(q13;q32) is the most common translocation, occurring in 20–25 % of all PCM. It involves the CCND1 gene and is associated with a good prognosis and long survival. PCM with the t(11;14)—CCND1–IGH translocation often show considerable morphologic and immunophenotypic overlap with LPL. Detection of the translocation and cyclin D1 protein expression by immunohistochemistry (IHC) are important in making the appropriate diagnosis [26].
The t(4;14)(p16.3;q32) portends the worst prognosis in PCM and is seen in approximately 25 % of cases; it involves IGH and the fibroblast growth factor receptor-3 (FGFR3) and MMSET genes at 4p16.3. The t(14;16)(q32;q23) translocation involving the MAF gene occurs in approximately 20 % of PCM and conveys an intermediate prognosis, as does the t(6;14)(p25;q32) translocation involving the MUM1 gene, also known as the IRF4 gene.
Progressive disease with transformation to high grade variants of PCM with large cell anaplastic or plasmablastic morphology is associated with MYC rearrangements [27, 28], TP53 mutations and deletions, mutations of KRAS and NRAS, 1p deletions, and amplifications of 1q21 [29, 30].
Cytogenetic abnormalities detected by karyotyping may show significantly different clinical outcomes compared to abnormalities detected by interphase FISH only [31]. For example, monosomy 13 detected by karyotyping is seen in approximately 50 % of PCM and is associated with poor survival, while chromosome 13 abnormalities detected by interphase FISH only are associated with intermediate survival. Multicolor FISH, also known as spectral karyotyping, can detect chromosomal abnormalities in PCM, including deletions, amplifications, and rearrangements that are missed or cryptic by conventional karyotyping, such as Xp11, 8q24, 11q13, 12q13, 13q21, 14q32, and 22q11.2 abnormalities [32]. New classification systems and stratification for treatment of PCM have been proposed due to these novel abnormalities, but further investigation into the clinical significance of this complex and expensive testing is needed before it can move into routine clinical care.
Marginal Zone B-Cell Lymphomas
The marginal zone lymphomas (MZL) are low grade B-cell lymphomas and include splenic marginal zone lymphoma (SMZL), extranodal marginal zone lymphomas of mucosa-associated lymphoid tissue (MALToma), and nodal marginal zone lymphoma (NMZL). Multiple molecular abnormalities occur in MZL, and MZL of different sites have different molecular abnormalities (Table 42.2). Testing for these abnormalities is helpful in confirmation of primary diagnosis and prognosis. Diagnostic and prognostic testing for these molecular abnormalities usually is performed by multicolor FISH.
The most common translocation in gastric, intestinal, and lung MALTomas is t(11;18)(q21;q21), which occurs in 30–50 % of cases [33–35]. The genes involved are the apoptosis inhibitor gene (API2) on 11q21, a member of the inhibitor of apoptosis (IAP) protein family with caspase-inhibitory functions, and the MALT1 gene on 18q21, encoding a human paracaspase protein. The resulting API2 – MALT1 chimeric transcript and fusion protein leads to inhibition of apoptosis and confers a survival advantage on the MZL cells. T(11;18)(q21;q21) is associated with worse prognosis and occurs most frequently in H. pylori-positive antibiotic-resistant MZL or advanced gastric MZL, but not in nodal or splenic MZL, nor in extranodal MZL with increased large cells or large-cell transformation. FISH is the most sensitive and specific method for detection of this translocation; however, reverse transcription PCR (RT-PCR) is occasionally used for detection.
An apoptosis regulatory molecule, BCL10, is overexpressed as a result of the t(1;14)(p22;q32) translocation, which involves BCL10 on 1p22 and the IGH gene on 14q32 [36]. Advanced MALTomas sometimes have both API2 – MALT1 and BCL10 – IGH translocations, and BCL10 appears to interact with the API2–MALT1 fusion protein to synergize activation of NFKB, suggesting that they are part of a common pathway. The BCL10 – IGH translocation is associated with a worse prognosis in low grade MALTomas and is typically detected by FISH.
The t(14;18)(q32;q21) translocation, also detected by FISH, involves IGH on 14q32 and MALT1 on 18q21. The IGH – MALT1 translocation occurs in most liver MALTomas, as well as some cutaneous, ocular adnexal, and salivary MALTomas, but is rare in MALTomas of the stomach, intestine, lung, thyroid, and breast, and does not occur in NMZL or SMZL. IGH – MALT1 does not usually occur with the API2 – MALT1 translocation, but sometimes occurs with trisomy 3 or trisomy 18 or both [37]. The clinical significance of this translocation is not yet established. This translocation should not be confused with the cytogenetically identical, but molecularly distinct t(14;18)(q32;q21) translocation, resulting in an IGH – BCL2 fusion, which is associated with follicular lymphoma.
Trisomy 3 occurs in more than 50 % of low-grade MZL, and aCGH studies have shown gains at 3q21-23 and 3q25-29, suggesting that BCL6 is involved in some cases. Trisomy 18 occurs in approximately 30 % of MZL. The genes involved are not well understood and the clinical relevance is unknown.
The t(3;14)(p13;q32) involving FOXP1 on chromosome 3 and the IGH gene on chromosome 14 is present in approximately 10 % of MALTomas. IGH – FOXP1 is seen most commonly in MZL arising in the thyroid, ocular adnexa, and skin and is absent in MZL from the lung and stomach. The translocation results in overexpression of FOXP1, is associated with trisomy 3, and may have a more aggressive clinical behavior [38, 39].
A20 (also known as TNFAIP3), found at 6q23, is a negative regulator of the NFKB pathway and is inactivated in a subset of MALTomas via mutation, methylation, or deletion [40]. A20 inactivation is preferentially seen in MALTomas arising from the ocular adnexa, salivary gland, and thyroid. Activating MYD88 point mutations (L265P), identical to those identified in the activated B-cell-like subtype of diffuse large B-cell lymphoma (DLBCL), and WM have been reported in 9 % of MALTomas, although the clinical significance is not yet clear [41]. Both A20 and MYD88 mutations contribute to constitutive activation of the NFKB pathway mediating cell survival and growth.
Follicular Lymphoma
Follicular lymphoma (FL) is the second most common subtype of NHL, accounting for approximately 22 % of cases [42]. FL patients demonstrate a remarkably variable clinical course, with some patients surviving for less than 1 year while others live for more than 20 years [43]. Standard pathologic, immunologic, and genetic analyses of FL have failed to identify a reliable method for prediction of prognosis in newly diagnosed FL patients. Gene expression profiling of a large series of FL suggests that the length of survival of FL patients correlates with the features of nonmalignant cells present in the lymphoma at diagnosis [44]. However, gene expression profiling is not available yet for routine clinical diagnosis and no alternative immunohistochemical algorithm has yet been developed in FL.
The t(14;18)(q32;q21) translocation, detected in approximately 80 % of FL at the time of initial diagnosis, is the cytogenetic hallmark of FL and juxtaposes the BCL2 oncogene on 18q21 with the IGH gene on 14q32. Overexpression of a normal BCL2 protein results from this translocation and protects the cells from apoptosis. Molecular analysis for IGH – BCL2 translocation using FISH (Fig. 42.2) is often used for confirmation of the initial diagnosis of FL in cases with equivocal histologic and immunologic findings. Molecular testing for IGH – BCL2 translocation by PCR in FL may be performed to evaluate the course of disease and the impact of therapy [45, 46]. Analysis to aid in evaluation of response to therapy is typically performed by PCR, because FISH is not sensitive enough for detection of a low level of MRD in BM or blood. BCL2 breakpoints occur most commonly in a 150 base pair (bp) span termed the major breakpoint cluster region (MBR), but may occur in areas of the gene called the minor breakpoint cluster region (MCR) or the intermediate cluster region (3′MBR). The insertion of variable numbers of random extra nucleotides at the breakpoint junction during crossover and the variability of the breakpoints in the BCL2 fusion gene and the six JH fragments results in considerable variation in the length of PCR products when testing is performed for this translocation. PCR fragment length usually ranges from 120 to 270 bp for the MBR, but occasional breakpoints as much as 800 bp downstream of the MBR region have been detected with standard MBR primers, resulting in PCR products of more than 1,000 bp. The clustering of breakpoints on chromosome 18 and the high degree of sequence homology among the 3′ portions of the JH segments make the IGH – BCL2 translocation very amenable to PCR detection and a good PCR target for MRD detection (see section on “Minimal Residual Disease Detection”).
Histologic progression occurs in approximately 30 % of FL patients, but the genetic events that are involved in this progression are not completely defined. Studies of paired lymph node biopsy specimens derived from the same FL patient before and after transformation identify at least two mechanisms for transformation. One is associated with high proliferation and one with recurrent oncogenic abnormalities. Mutations in TP53 occur in 20 % of transformed FL. MYC gene expression is markedly increased in 70 % of transformed FL, but appears to be a surrogate marker for the gene expression profiling high proliferation signature [47]. The frequency of MYC mutations and translocations in transformed FL is low [48]. Copy number variability in two regions (1p36.22-p36.33 and 6q21-q24.3) are predictors of transformation risk in FL, independent of the International Prognostic Index (IPI) score. These prognostic features may be useful to identify high-risk patients as candidates for risk-adapted therapies [49].
Genes encoding proteins involved with histone modification are frequent targets of somatic hypermutation in both DLBCL and FL and may play an important role in B-cell lymphomagenesis. Both DLBCL and FL may have mutations in these genes, but the frequency of mutation is highest in FL, with MLL2 mutation in 89 % and MEF2B mutation in 13 % of FL [50]. MLL2 encodes a histone methyltransferase and MEF2b cooperates with CREBBP and EP300 to acetylate histones.
Mantle Cell Lymphoma
Molecular testing is performed at initial diagnosis in most patients with MCL as the diagnosis confers a very poor prognosis and mandates aggressive therapy in most cases [51]. Diagnostic testing targets the t(11;14)(q13;q32) translocation, which juxtaposes the BCL1 or CCND1 gene on chromosome 11q13 with an IGH enhancer, resulting in overexpression of normal cyclin D1 protein and increased cell cycling. This translocation is not completely specific for MCL, as it also occurs in many plasma cell myelomas. However, no diagnostic problems are caused by this overlap as the CCND1 – IGH translocation does not occur in the subtypes of BCL with which MCL shows significant morphologic or immunophenotypic overlap. Multiple methods have been used to detect the CCND1 – IGH translocation, including karyotyping, PCR, RT-PCR for CCND1 mRNA, and FISH [52]. The optimal diagnostic method has proven to be FISH (Fig. 42.3), which detects > 90 % of cases, even in formalin-fixed paraffin-embedded (FFPE) biopsy tissue. PCR detection is less successful because the 11q13 breakpoints are widely distributed; approximately 30–50 % are localized to a 1 kb DNA segment called the major translocation cluster (MTC), but the remaining translocations involve many different sites not easily detectable by PCR analysis. There is little application for the PCR assay when FISH is faster, more specific, and can be performed on FFPE tissue.
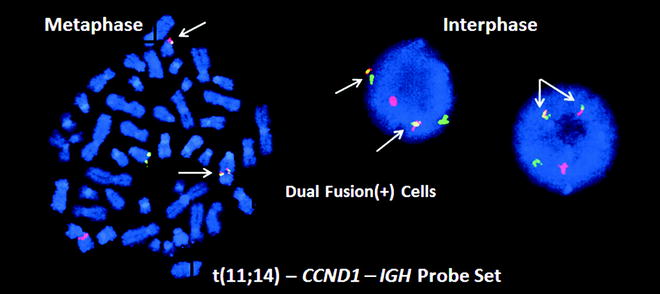
Figure 42.3
FISH analysis for the t(11;14) CCND1 – IGH translocation in a mantle cell lymphoma (MCL). On the left is a single metaphase spread and on the right are two interphase cells, all with two yellow, one red and one green signal called a dual fusion signal pattern (RGYY). These probes detect both derivative chromosomes resulting from the translocation. Both derivative chromosomes produce a yellow fusion signal, whereas the normal alleles produce separate red and green signals
The proliferation signature by gene expression profiling can identify MCL patients with the poorest prognosis [53]. The evaluation of Ki-67 by IHC is often used as a surrogate for the gene expression proliferation signature in the routine diagnostic workup of MCL, but is difficult to interpret and is less predictive in the intermediate range. However, a high level of Ki-67 expression is likely associated with poor outcome, independent of the morphologic variant. The mitotic index (counting the number of mitotic figures/mm2) may be superior to Ki-67 expression [54]. Gene expression profiling has identified the transcription factor SOX11 as a specific marker for MCL and IHC analysis demonstrates strong nuclear staining with SOX11 in > 90 % of MCL cases including those that are cyclin D1-negative [55]. While this is potentially a diagnostically useful marker, the prognostic significance remains controversial.
ATM deletions like those in CLL/SLL also are seen in many MCL cases, and progression or aggressive clinical behavior in MCL is associated with P16 and TP53 abnormalities [56]. Additionally, recurrent NOTCH1 mutations occur in MCL; similarly to CLL/SLL, NOTCH1 mutations are associated with poor prognosis [57]. MCLs rarely have somatic hypermutation of IGVH.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common subtype of lymphoma and this category of the WHO classification represents another clinically and biologically heterogeneous group of BCL. Over the past decade, promising new technologies have been applied to the problem of identification of prognostic subgroups of DLBCL. Microarray gene expression profiling has identified three molecularly and prognostically distinct subtypes of DLBCL: a germinal center B-cell-like DLBCL subgroup (GCB) with a favorable prognosis; an activated B-cell-like DLBCL subgroup (ABC) with a poorer prognosis; and a favorable primary mediastinal large B-cell lymphoma subgroup (PMBL) [58–60].
In addition to distinct gene expression patterns, these DLBCL subgroups have different cytogenetic and molecular abnormalities and utilize different oncogenic pathways. The GCB type of DLBCL derives from the germinal center B cell. IGH – BCL2 translocations [t(14;18)] identical to those seen in FL are specific to GCB DLBCL and are not seen in the other DLBCL subtypes. GCB DLBCL may also have amplification of the REL locus on chromosome 2p, deletion of PTEN on chromosome 10, and amplification of microRNA cluster mir-17-92 [61], as well as mutations in genes involved in chromatin/histone modification such as EZH2 and CREBBP [50, 62] (Table 42.3).
ABC DLBCL originates from a post germinal center B cell, has a gene expression profile similar to that of activated peripheral blood B cells [58], and is characterized by constitutive activation of the NFKB pathway. Mutations in A20, MYD88 and CARD11 are thought to contribute to deregulation of the NFKB pathway [63]. Also typical of ABC DLBCL are trisomy 3, increased FOXP1 mRNA expression, deletion of chromosome 6q, deletion of the INK4a – ARF tumor suppressor locus on chromosome 9, and gain or amplification of part of chromosome 19 [61] (Table 42.3).
PMBL represents a distinct entity in the 2008 WHO classification, which is more frequent in young adults and typically presents as a mediastinal mass without involvement of lymph nodes or BM. PMBL is thought to be derived from a thymic medullary B cell. The gene expression profile more closely resembles that of classical Hodgkin lymphoma than the other subtypes of DLBCL. aCGH has demonstrated amplification of chromosome 9p24 in up to 75 % of cases and 2p15 in approximately 50 % of cases. Candidate genes include REL and BCL11A at 2p and JAK2, PDL1, and PDL2 at 9p [64]. SOCS1 mutations and upregulation of NFKB are also genetic hallmarks of PMBL [65].
Mutations or translocations in BCL6 (3q27) are common abnormalities in DLBCL, occurring in approximately 30 % of cases and not associated with a specific subgroup [66, 67]. Translocations involve both IGH and non IGH gene partners. The particular translocation partner may dictate the prognostic significance of the BCL6 translocation, with translocations involving the IGH locus having a more favorable prognosis than those with other partner genes. MYC translocations are present in approximately 10 % of DLBCL. In contrast to BL, MYC rearrangements in DLBCL often are present in the context of a complex karyotype, and in 30–40 % of cases, MYC is rearranged with a non-Ig gene. FISH using a MYC break-apart probe is useful in the detection of these cases (Fig. 42.2). MYC translocations in DLBCL have a worse prognosis with poor response to therapy [68, 69]. Of note, FISH analysis does not detect all cases with significant expression of MYC. IHC for MYC protein identifies a subset of MYC FISH-negative DLBCL cases with high levels of MYC protein expression. These IHC-positive, FISH-negative cases have a very poor clinical prognosis, but only if there is also high expression of BCL2 protein [70, 71].
Burkitt Lymphoma
BL is a high grade BCL that is characteristically associated with the t(8;14)(q24;q32) translocation involving the MYC gene on 8q24 and the IGH gene on 14q32 [72]. Occasional BL demonstrates variant t(2;8)(p11;q24) or t(8;22)(q24;q11) translocations involving MYC and the kappa and lambda light chain loci, respectively [73]. These translocations result in increased expression of MYC and markedly increased cell proliferation. The site of translocation of MYC is reported to vary between sporadic and endemic BL. In endemic disease, the 8q24 breakpoint occurs up to 300 kb 5′ from the coding region of the MYC gene, while sporadic BL characteristically involves a breakpoint in MYC exon 1 [74]. The site of translocation in the IGH gene also is variable, with endemic cases involving the J region and sporadic cases involving the C regions. The degree of molecular variability in these translocations is the reason they are not amenable to detection by standard PCR assays. Routine karyotyping and FISH analysis using MYC probes are successful in detecting almost all MYC translocations in BL. BL typically do not have other chromosomal abnormalities. Gene expression profiling of BL and DLBCL has identified typical gene signatures for BL vs DLBCL [75]. The MYC protein and its target genes, as well as some genes expressed in normal germinal center B cells are more highly expressed in BL than GCB DLBCL, while major histocompatibility complex (MHC) class I genes and NFKB target genes are expressed at lower levels in BL than in DLBCL. Some of the recurrently mutated genes in DLBCL (EZH2, SGK1, BCL2, CD79B, and MYD88) are rarely mutated in BL [76].
B-Cell Lymphoma, Unclassifiable, with Features Intermediate Between DLBCL and BL
A subset of B-cell lymphomas shows morphologic and immunologic overlap with both BL and DLBCL. In some but not all of these cases, the gene expression profile is similar to BL [75]. Historically, these have been called “atypical Burkitt” or “Burkitt-like” lymphomas, but in the 2008 WHO classification, they were put into the category of BCL, unclassifiable, with features intermediate between DLBCL and BL (BCLU) [1]. Unlike classical BL, MYC translocations in BCLU frequently involve a non-Ig gene partner and may occur in the background of a complex karyotype. Most of the so-called “double hit lymphomas,” defined by the presence of a MYC rearrangement and another genetic abnormality, most commonly BCL2 gene rearrangement, are included in this category. However, some double hit lymphomas may be morphologically and phenotypically indistinguishable from DLBCL or even FL. Identification of double hit lymphomas at diagnosis is clinically important because, regardless of how they are subclassified morphologically, double hit lymphomas have a particularly aggressive clinical course and are often refractory to conventional chemotherapeutic regimens used for DLBCL. In rare cases of “triple hit lymphoma,” MYC, BCL2, and BCL6 are all rearranged and the clinical outcome is particularly dismal. FISH analysis is typically used at diagnosis for detection of these cytogenetic abnormalities, but will not detect all cases, particularly those with other mechanisms of overexpression of MYC. Because of the possible overlap with typical DLBCL, some institutions now routinely use a diagnostic FISH panel for detection of MYC rearrangements plus IGH – BCL2 and BCL6 rearrangements at the time of initial diagnosis for all new DLBCLs and BCLUs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


