49
Antiviral Agents
CASE STUDY
A 35-year-old white woman who recently tested seropositive for both HIV and hepatitis B virus surface antigen is referred for evaluation. She is feeling well overall but reports a 25-pack-year smoking history. She drinks 3-4 beers per week and has no known medication allergies. She has a history of heroin use and is currently receiving methadone. Physical examination reveals normal vital signs and no abnormalities. White blood cell count is 5800 cells/mm3 with a normal differential, hemoglobin is 11.8 g/dL, all liver tests are within normal limits, CD4 cell count is 278 cells/mm3, and viral load (HIV RNA) is 110,000 copies/mL. What other laboratory tests should be ordered? Which antiretroviral medications would you begin?
Viruses are obligate intracellular parasites; their replication depends primarily on synthetic processes of the host cell. Therefore, to be effective, antiviral agents must either block viral entry into or exit from the cell or be active inside the host cell. As a corollary, nonselective inhibitors of virus replication may interfere with host cell function and result in toxicity.
Progress in antiviral chemotherapy began in the early 1950s, when the search for anti-cancer drugs generated several new compounds capable of inhibiting viral DNA synthesis. The two first-generation antiviral agents, 5-iododeoxyuridine and trifluorothymidine, had poor specificity (ie, they inhibited host cell DNA as well as viral DNA) that rendered them too toxic for systemic use. However, both agents are effective when used topically for the treatment of herpes keratitis.
Knowledge of the mechanisms of viral replication has provided insights into critical steps in the viral life cycle that can serve as potential targets for antiviral therapy. Recent research has focused on identifying agents with greater selectivity, higher potency, in vivo stability, and reduced toxicity. Antiviral therapy is now available for herpesviruses, hepatitis C virus (HCV), hepatitis B virus (HBV), papillomavirus, influenza, human immunodeficiency virus (HIV), and respiratory syncytial virus (RSV). Antiviral drugs share the common property of being virustatic; they are active only against replicating viruses and do not affect latent virus. Whereas some infections require monotherapy for brief periods of time (eg, acyclovir for herpes simplex virus), others require dual therapy for prolonged periods of time (interferon alfa/ribavirin for HCV), whereas still others require multiple drug therapy for indefinite periods (HIV). In chronic illnesses such as viral hepatitis and HIV infection, potent inhibition of viral replication is crucial in limiting the extent of systemic damage.
ACRONYMS & OTHER NAMES
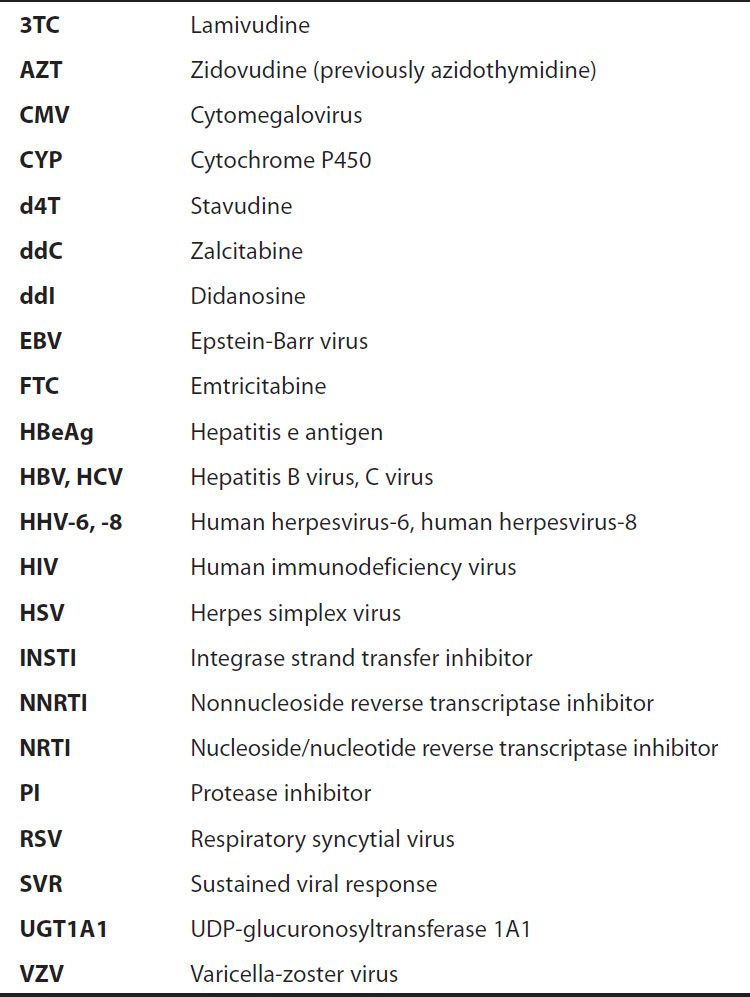
Viral replication requires several steps (Figure 49–1): (1) attachment of the virus to receptors on the host cell surface; (2) entry of the virus through the host cell membrane; (3) uncoating of viral nucleic acid; (4) synthesis of early regulatory proteins, eg, nucleic acid polymerases; (5) synthesis of new viral RNA or DNA; (6) integration into the nuclear genome; (7) synthesis of late, structural proteins; (8) assembly (maturation) of viral particles; and (9) release from the cell. Antiviral agents can potentially target any of these steps.
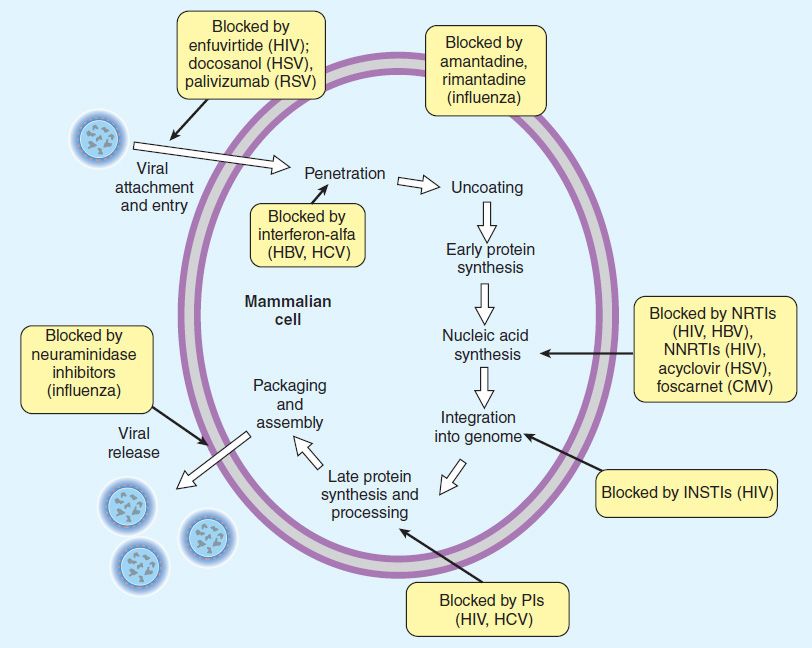
FIGURE 49–1 The major sites of antiviral drug action. Note: Interferon alfas are speculated to have multiple sites of action. (Modified and reproduced, with permission, from Trevor AJ, Katzung BG, Masters SB: Pharmacology: Examination & Board Review, 9th ed. McGraw-Hill, 2010. Copyright © The McGraw-Hill Companies, Inc.)
 AGENTS TO TREAT HERPES SIMPLEX VIRUS (HSV) & VARICELLA-ZOSTER VIRUS (VZV) INFECTIONS
AGENTS TO TREAT HERPES SIMPLEX VIRUS (HSV) & VARICELLA-ZOSTER VIRUS (VZV) INFECTIONS
Three oral nucleoside analogs are licensed for the treatment of HSV and VZV infections: acyclovir, valacyclovir, and famciclovir. They have similar mechanisms of action and comparable indications for clinical use; all are well tolerated. Acyclovir has been the most extensively studied; it was licensed first and is the only one of the three that is available for intravenous use in the United States. Comparative trials have demonstrated similar efficacies of these three agents for the treatment of HSV but modest superiority of famciclovir and valacyclovir for the treatment of herpes zoster infections.
ACYCLOVIR
Acyclovir (eFigure 49–1.1) is an acyclic guanosine derivative with clinical activity against HSV-1, HSV-2, and VZV, but it is approximately 10 times more potent against HSV-1 and HSV-2 than against VZV. In vitro activity against Epstein-Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus-6 (HHV-6) is present but weaker.
Acyclovir requires three phosphorylation steps for activation. It is converted first to the monophosphate derivative by the virus-specified thymidine kinase and then to the di- and triphosphate compounds by host cell enzymes (Figure 49–2). Because it requires the viral kinase for initial phosphorylation, acyclovir is selectively activated—and the active metabolite accumulates—only in infected cells. Acyclovir triphosphate inhibits viral DNA synthesis by two mechanisms: competition with deoxyGTP for the viral DNA polymerase, resulting in binding to the DNA template as an irreversible complex; and chain termination following incorporation into the viral DNA.
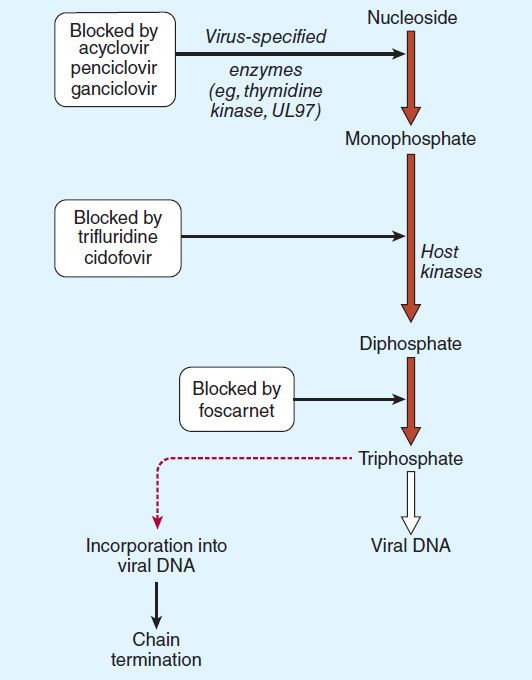
FIGURE 49–2 Mechanism of action of antiherpes agents.
The bioavailability of oral acyclovir is low (15–20%) and is unaffected by food. An intravenous formulation is available. Topical formulations produce high concentrations in herpetic lesions, but systemic concentrations are undetectable by this route.
Acyclovir is cleared primarily by glomerular filtration and tubular secretion. The half-life is 2.5–3 hours in patients with normal renal function and 20 hours in patients with anuria. Acyclovir diffuses readily into most tissues and body fluids. Cerebrospinal fluid concentrations are 20–50% of serum values.
Oral acyclovir has multiple uses. In first episodes of genital herpes, oral acyclovir shortens the duration of symptoms by approximately 2 days, the time to lesion healing by 4 days, and the duration of viral shedding by 7 days. In recurrent anogenital herpes, the time course is shortened by 1–2 days. Treatment of first-episode genital herpes does not alter the frequency or severity of recurrent outbreaks. Long-term suppression with oral acyclovir in patients with frequent recurrences of genital herpes decreases the frequency of symptomatic recurrences and of asymptomatic viral shedding, thus decreasing the rate of sexual transmission. However, outbreaks may resume upon discontinuation of suppressive acyclovir. Oral acyclovir is only modestly beneficial in recurrent herpes labialis. In contrast, acyclovir therapy significantly decreases the total number of lesions, duration of symptoms, and viral shedding in patients with varicella (if begun within 24 hours after the onset of rash) or cutaneous zoster (if begun within 72 hours); the risk of post-herpetic neuralgia is also reduced if treatment is initiated early. However, because VZV is less susceptible to acyclovir than HSV, higher doses are required (Table 49–1). When given prophylactically to patients undergoing organ transplantation, oral or intravenous acyclovir prevents reactivation of HSV and VZV infection. Evidence from clinical trials suggests that the use of daily acyclovir (400 mg twice daily) may reduce the plasma viral load of HIV-1 and the risk of HIV-associated disease progression in individuals dually infected with HSV-2 and HIV-1.
TABLE 49–1 Agents to treat or prevent herpes simplex virus (HSV) and varicella-zoster virus (VZV) infections.
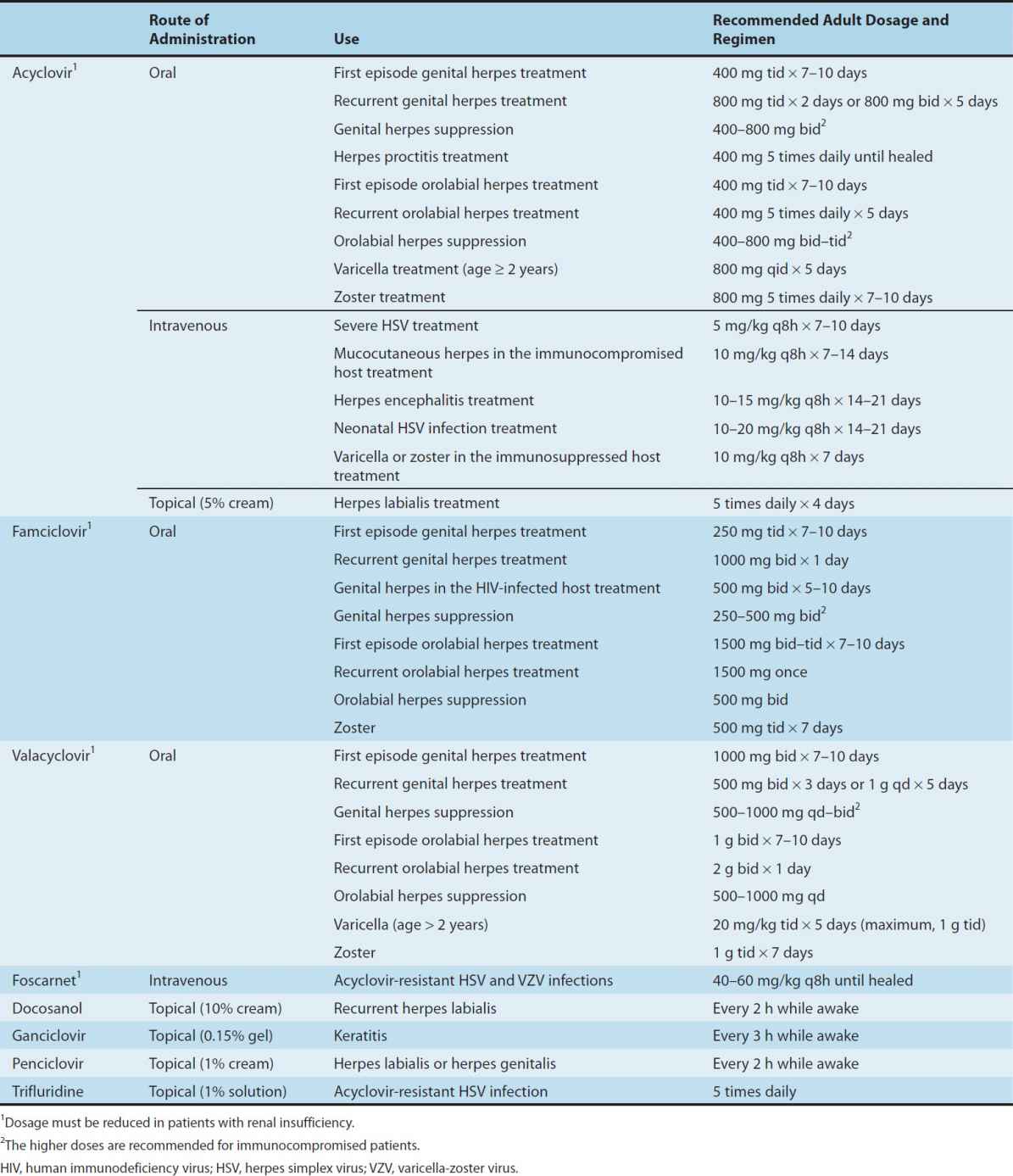
Intravenous acyclovir is the treatment of choice for herpes simplex encephalitis, neonatal HSV infection, and serious HSV or VZV infections (Table 49–1). In neonates with central nervous system HSV, oral acyclovir suppression for 6 months following acute treatment improves neurodevelopmental outcomes. In immunocompromised patients with VZV infection, intravenous acyclovir reduces the incidence of cutaneous and visceral dissemination.
Topical acyclovir cream is substantially less effective than oral therapy for primary HSV infection. It is of no benefit in treating recurrent genital herpes.
Resistance to acyclovir can develop in HSV or VZV through alteration in either the viral thymidine kinase or the DNA polymerase, and clinically resistant infections have been reported in immunocompromised hosts. Most clinical isolates are resistant on the basis of deficient thymidine kinase activity and thus are cross-resistant to valacyclovir, famciclovir, and ganciclovir. Agents such as foscarnet, cidofovir, and trifluridine do not require activation by viral thymidine kinase and thus have preserved activity against the most prevalent acyclovir-resistant strains (Figure 49–2).
Acyclovir is generally well tolerated, although nausea, diarrhea, and headache may occur. Intravenous infusion may be associated with reversible renal toxicity (ie, crystalline nephropathy or interstitial nephritis) or neurologic effects (eg, tremors, delirium, seizures). However, these are uncommon with adequate hydration and avoidance of rapid infusion rates. High doses of acyclovir cause chromosomal damage and testicular atrophy in rats, but there has been no evidence of teratogenicity, reduction in sperm production, or cytogenetic alterations in peripheral blood lymphocytes in patients receiving daily suppression of genital herpes for more than 10 years. A recent study found no evidence of increased birth defects in 1150 infants who were exposed to acyclovir during the first trimester. In fact, the American College of Obstetricians and Gynecologists recommends suppressive acyclovir therapy beginning at week 36 in pregnant women with active recurrent genital herpes to reduce the risk of recurrence at delivery and possibly the need for cesarean section. The impact of this intervention on neonatal infection has not been established.
Concurrent use of nephrotoxic agents may enhance the potential for nephrotoxicity. Probenecid and cimetidine decrease acyclovir clearance and increase exposure. Somnolence and lethargy may occur in patients receiving concomitant zidovudine and acyclovir.
VALACYCLOVIR
Valacyclovir is the L-valyl ester of acyclovir. It is rapidly converted to acyclovir after oral administration via first-pass enzymatic hydrolysis in the liver and intestine, resulting in serum levels that are three to five times greater than those achieved with oral acyclovir and approximate those achieved with intravenous acyclovir. Oral bioavailability is 54–70%, and cerebrospinal fluid levels are about 50% of those in serum. Elimination half-life is 2.5–3.3 hours.
Twice-daily valacyclovir is effective for treatment of first or recurrent genital herpes and varicella and zoster infections; it is approved for use as a 1-day treatment for orolabial herpes and as suppression of frequently recurring genital herpes (Table 49–1). Once-daily dosing of valacyclovir for chronic suppression in persons with recurrent genital herpes has been shown to markedly decrease the risk of sexual transmission. In comparative trials with acyclovir for the treatment of patients with zoster, rates of cutaneous healing were similar, but valacyclovir was associated with a shorter duration of zoster-associated pain. Higher doses of valacyclovir (2 g four times daily) are effective in preventing CMV disease after organ transplantation and suppressive valacyclovir prevents VZV reactivation after hematopoietic stem cell transplantation.
Valacyclovir is generally well tolerated, although nausea, headache, vomiting, or rash may occur. At high doses, confusion, hallucinations, and seizures have been reported. AIDS patients who received high-dosage valacyclovir chronically (ie, 8 g/d) had increased gastrointestinal intolerance as well as thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome; this dose has also been associated with confusion and hallucinations in transplant patients. In a recent study, there was no evidence of increased birth defects in 181 infants who were exposed to valacyclovir during the first trimester.
FAMCICLOVIR
Famciclovir is the diacetyl ester prodrug of 6-deoxypenciclovir, an acyclic guanosine analog (eFigure 49–2.1). After oral administration, famciclovir is rapidly deacetylated and oxidized by first-pass metabolism to penciclovir. It is active in vitro against HSV-1, HSV-2, VZV, EBV, and HBV. As with acyclovir, activation by phosphorylation is catalyzed by the virus-specified thymidine kinase in infected cells, followed by competitive inhibition of the viral DNA polymerase to block DNA synthesis. Unlike acyclovir, however, penciclovir does not cause chain termination. Penciclovir triphosphate has lower affinity for the viral DNA polymerase than acyclovir triphosphate, but it achieves higher intracellular concentrations. The most commonly encountered clinical mutants of HSV are thymidine kinase-deficient; these are cross-resistant to acyclovir and famciclovir.
The bioavailability of penciclovir from orally administered famciclovir is 70%. The intracellular half-life of penciclovir triphosphate is prolonged, at 7–20 hours. Penciclovir is excreted primarily in the urine.
Oral famciclovir is effective for the treatment of first and recurrent genital herpes, for chronic daily suppression of genital herpes, for treatment of herpes labialis, and for the treatment of acute zoster (Table 49–1). One-day usage of famciclovir significantly accelerates time to healing of recurrent genital herpes and of herpes labialis. Comparison of famciclovir to valacyclovir for treatment of herpes zoster in immunocompetent patients showed similar rates of cutaneous healing and pain resolution; both agents shortened the duration of zoster-associated pain compared with acyclovir.
Oral famciclovir is generally well tolerated, although headache, nausea, or diarrhea may occur. As with acyclovir, testicular toxicity has been demonstrated in animals receiving repeated doses. However, men receiving daily famciclovir (250 mg every 12 hours) for 18 weeks had no changes in sperm morphology or motility. In a recent study, there was no evidence of increased birth defects in 32 infants who were exposed to famciclovir during the first trimester. The incidence of mammary adenocarcinoma was increased in female rats receiving famciclovir for 2 years.
PENCICLOVIR
The guanosine analog penciclovir, the active metabolite of famciclovir, is available for topical use. Penciclovir cream (1%) shortened the median duration of recurrent herpes labialis by ~ 17 hours compared to placebo when applied within 1 hour of the onset of prodromal symptoms and continued every 2 hours during waking hours for 4 days. Adverse effects are uncommon, other than application site reactions in ~1%.
DOCOSANOL
Docosanol is a saturated 22-carbon aliphatic alcohol that inhibits fusion between the host cell plasma membrane and the HSV envelope, thereby preventing viral entry into cells and subsequent viral replication. Topical docosanol 10% cream is available without a prescription. When applied within 12 hours of the onset of prodromal symptoms, five times daily, median healing time was shortened by 18 hours compared with placebo in recurrent orolabial herpes. Application site reactions occur in ~2%.
TRIFLURIDINE
Trifluridine (trifluorothymidine) is a fluorinated pyrimidine nucleoside that inhibits viral DNA synthesis in HSV-1, HSV-2, CMV, vaccinia, and some adenoviruses. It is phosphorylated intracellularly by host cell enzymes, and then competes with thymidine triphosphate for incorporation by the viral DNA polymerase (Figure 49–2). Incorporation of trifluridine triphosphate into both viral and host DNA prevents its systemic use. Application of a 1% solution is effective in treating keratoconjunctivitis and recurrent epithelial keratitis due to HSV-1 or HSV-2. Cutaneous application of trifluridine solution, alone or in combination with interferon alfa, has been used successfully in the treatment of acyclovir-resistant HSV infections.
INVESTIGATIONAL AGENTS
Valomaciclovir is an inhibitor of the viral DNA polymerase; it is currently under clinical evaluation for the treatment of patients with acute zoster and acute EBV infection (infectious mononucleosis).
 AGENTS TO TREAT CYTOMEGALOVIRUS (CMV) INFECTIONS
AGENTS TO TREAT CYTOMEGALOVIRUS (CMV) INFECTIONS
CMV infections occur primarily in the setting of advanced immunosuppression and are typically due to reactivation of latent infection. Dissemination of infection results in end-organ disease, including retinitis, colitis, esophagitis, central nervous system disease, and pneumonitis. Although the incidence in HIV-infected patients has markedly decreased with the advent of potent anti-retroviral therapy, clinical reactivation of CMV infection after organ transplantation is still prevalent.
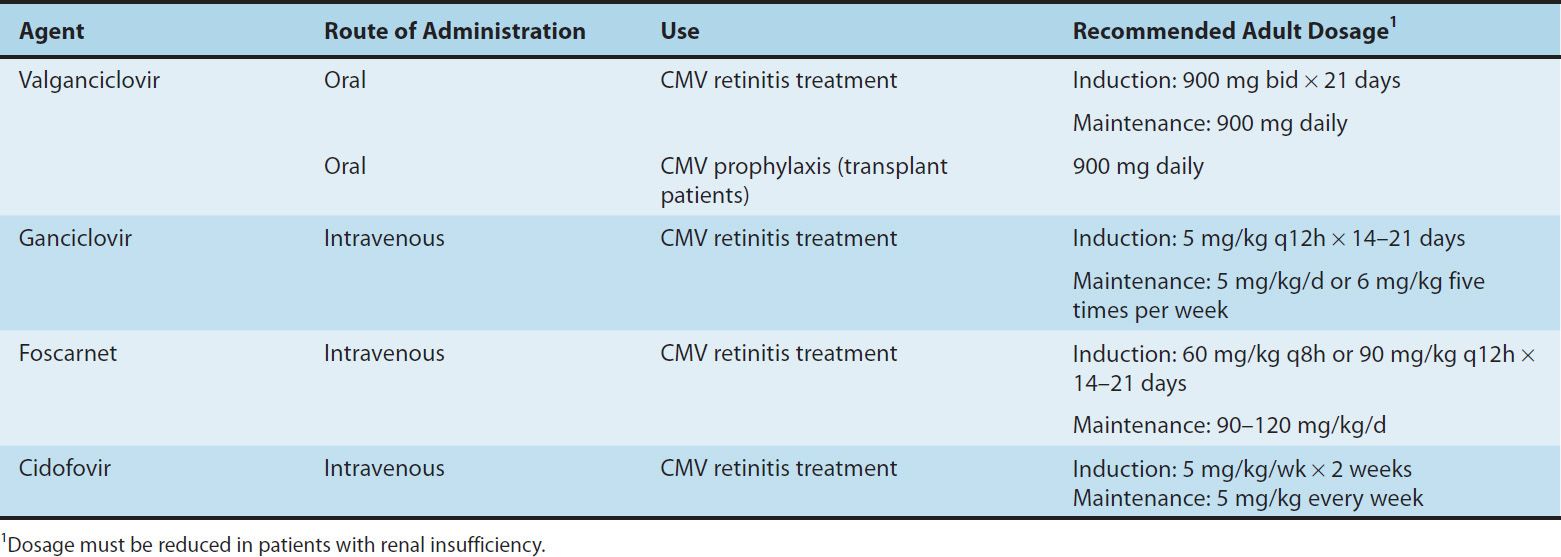
The availability of oral valganciclovir has decreased the use of intravenous ganciclovir, intravenous foscarnet, and intravenous cidofovir for the prophylaxis and treatment of end-organ CMV disease (Table 49–2). Oral valganciclovir has replaced oral ganciclovir because of its lower pill burden.
TABLE 49–2 Agents to treat cytomegalovirus (CMV) infection.
GANCICLOVIR
Ganciclovir is an acyclic guanosine analog (eFigure 49–2.1) that requires activation by triphosphorylation before inhibiting the viral DNA polymerase. Initial phosphorylation is catalyzed by the virus-specified protein kinase phosphotransferase UL97 in CMV-infected cells. The activated compound competitively inhibits viral DNA polymerase and causes termination of viral DNA elongation (Figure 49–2). Ganciclovir has in vitro activity against CMV, HSV, VZV, EBV, HHV-6, and HHV-8. Its activity against CMV is up to 100 times greater than that of acyclovir.
Ganciclovir is administered intravenously; the bioavailability of oral ganciclovir is poor, and it is no longer available in the US. Ganciclovir gel is available for the treatment of acute herpetic keratitis. Cerebrospinal fluid concentrations are approximately 50% of serum concentrations. The elimination half-life is 4 hours, and the intracellular half-life is prolonged at 16–24 hours. Clearance of the drug is linearly related to creatinine clearance. Ganciclovir is readily cleared by hemodialysis.
Intravenous ganciclovir has been shown to delay progression of CMV retinitis in immunocompromised patients. Dual therapy with foscarnet and ganciclovir is more effective in delaying progression of retinitis than either drug alone in patients with AIDS (see Foscarnet), although adverse effects are compounded. Intravenous ganciclovir is also used to treat CMV colitis, esophagitis, and pneumonitis (the latter often in combination with intravenous cytomegalovirus immunoglobulin) in immunocompromised patients. Intravenous ganciclovir, followed by either oral ganciclovir or high-dose oral acyclovir, reduced the risk of CMV infection in transplant recipients. Limited data in infants with symptomatic congenital neurologic CMV disease suggest that treatment with IV ganciclovir may reduce hearing loss. The risk of Kaposi’s sarcoma is reduced in AIDS patients receiving long-term ganciclovir, presumably because of activity against HHV-8.
Intravitreal injections of ganciclovir may be used to treat CMV retinitis. Concurrent therapy with a systemic anti-CMV agent is necessary to prevent other sites of end-organ CMV disease. The intraocular ganciclovir implant is no longer available in the USA.
Resistance to ganciclovir increases with duration of use. The more common mutation, in UL97, results in decreased levels of the triphosphorylated (ie, active) form of ganciclovir. The less common UL54 mutation in DNA polymerase results in higher levels of resistance and potential cross-resistance with cidofovir and foscarnet. Antiviral susceptibility testing is recommended in patients in whom resistance is suspected clinically.
The most common adverse effect of intravenous ganciclovir treatment is myelosuppression, which although reversible may be dose-limiting. Myelosuppression may be additive in patients receiving concurrent zidovudine, azathioprine, or mycophenolate mofetil. Other potential adverse effects are nausea, diarrhea, fever, rash, headache, insomnia, and peripheral neuropathy. Central nervous system toxicity (confusion, seizures, psychiatric disturbance) and hepatotoxicity have been rarely reported. Intravitreal ganciclovir has been associated with vitreous hemorrhage and retinal detachment. Ganciclovir is mutagenic in mammalian cells and carcinogenic and embryotoxic at high doses in animals and causes aspermatogenesis; the clinical significance of these preclinical data is unclear.
Levels of ganciclovir may rise in patients concurrently taking probenecid or trimethoprim. Concurrent use of ganciclovir with didanosine may result in increased levels of didanosine.
VALGANCICLOVIR
Valganciclovir is an L-valyl ester prodrug of ganciclovir that exists as a mixture of two diastereomers. After oral administration, both diastereomers are rapidly hydrolyzed to ganciclovir by esterases in the intestinal wall and liver.
Valganciclovir is well absorbed; the bioavailability of oral valganciclovir is 60% and it is recommended that the drug be taken with food. The AUC0–24h resulting from valganciclovir (900 mg once daily) is similar to that after 5 mg/kg once daily of intravenous ganciclovir and approximately 1.65 times that of oral ganciclovir. The major route of elimination is renal, through glomerular filtration and active tubular secretion. Plasma concentrations of valganciclovir are reduced approximately 50% by hemodialysis.
Valganciclovir is as effective as intravenous ganciclovir for the treatment of CMV retinitis and is also indicated for the prevention of CMV disease in high-risk solid organ and bone marrow transplant recipients. Adverse effects, drug interactions, and resistance patterns are the same as those associated with ganciclovir.
FOSCARNET
Foscarnet (phosphonoformic acid) is an inorganic pyrophosphate analog that inhibits herpesvirus DNA polymerase, RNA polymerase, and HIV reverse transcriptase directly without requiring activation by phosphorylation. Foscarnet blocks the pyrophosphate binding site of these enzymes and inhibits cleavage of pyrophosphate from deoxynucleotide triphosphates. It has in vitro activity against HSV, VZV, CMV, EBV, HHV-6, HHV-8, HIV-1, and HIV-2.
Foscarnet is available in an intravenous formulation only; poor oral bioavailability and gastrointestinal intolerance preclude oral use. Cerebrospinal fluid concentrations are 43–67% of steady-state serum concentrations. Although the mean plasma half-life is 3–7 hours, up to 30% of foscarnet may be deposited in bone, with a half-life of several months. The clinical repercussions of this are unknown. Clearance of foscarnet is primarily renal and is directly proportional to creatinine clearance. Serum drug concentrations are reduced approximately 50% by hemodialysis.
Foscarnet is effective in the treatment of end-organ CMV disease (ie, retinitis, colitis, and esophagitis), including ganciclovir-resistant disease; it is also effective against acyclovir-resistant HSV and VZV infections. The dosage of foscarnet must be titrated according to the patient’s calculated creatinine clearance before each infusion. Use of an infusion pump to control the rate of infusion is important to prevent toxicity, and large volumes of fluid are required because of the drug’s poor solubility. The combination of ganciclovir and foscarnet is synergistic in vitro against CMV and has been shown to be superior to either agent alone in delaying progression of retinitis; however, toxicity is also increased when these agents are administered concurrently. As with ganciclovir, a decrease in the incidence of Kaposi’s sarcoma has been observed in patients who have received long-term foscarnet.
Foscarnet has been administered intravitreally for the treatment of CMV retinitis in patients with AIDS, but data regarding efficacy and safety are incomplete.
Resistance to foscarnet in HSV and CMV isolates is due to point mutations in the DNA polymerase gene and is typically associated with prolonged or repeated exposure to the drug. Mutations in the HIV-1 reverse transcriptase gene have also been described. Although foscarnet-resistant CMV isolates are typically cross-resistant to ganciclovir, foscarnet activity is usually maintained against ganciclovir- and cidofovir-resistant isolates of CMV.
Potential adverse effects of foscarnet include renal impairment, hypo- or hypercalcemia, hypo- or hyperphosphatemia, hypokalemia, and hypomagnesemia. Saline preloading helps prevent nephrotoxicity, as does avoidance of concomitant administration of drugs with nephrotoxic potential (eg, amphotericin B, pentamidine, aminoglycosides). The risk of severe hypocalcemia, caused by chelation of divalent cations, is increased with concomitant use of pentamidine. Genital ulcerations associated with foscarnet therapy may be due to high levels of ionized drug in the urine. Nausea, vomiting, anemia, elevation of liver enzymes, and fatigue have been reported; the risk of anemia may be additive in patients receiving concurrent zidovudine. Central nervous system toxicity includes headache, hallucinations, and seizures; the risk of seizures may be increased with concurrent use of imipenem. Foscarnet caused chromosomal damage in preclinical studies.
CIDOFOVIR
Cidofovir (eFigure 49–2.1) is a cytosine nucleotide analog with in vitro activity against CMV, HSV-1, HSV-2, VZV, EBV, HHV-6, HHV-8, adenovirus, poxviruses, polyomaviruses, and human papillomavirus. In contrast to ganciclovir, phosphorylation of cidofovir to the active diphosphate is independent of viral enzymes (Figure 49–2); thus activity is maintained against thymidine kinase-deficient or -altered strains of CMV or HSV. Cidofovir diphosphate acts both as a potent inhibitor of and as an alternative substrate for viral DNA polymerase, competitively inhibiting DNA synthesis and becoming incorporated into the viral DNA chain. Cidofovir-resistant isolates tend to be cross-resistant with ganciclovir but retain susceptibility to foscarnet.
Although the terminal half-life of cidofovir is approximately 2.6 hours, the active metabolite cidofovir diphosphate has a prolonged intracellular half-life of 17–65 hours, thus allowing infrequent dosing. A separate metabolite, cidofovir phosphocholine, has a half-life of at least 87 hours and may serve as an intracellular reservoir of active drug. Cerebrospinal fluid penetration is poor. Elimination is by active renal tubular secretion. High-flux hemodialysis reduces serum levels of cidofovir by approximately 75%.
Intravenous cidofovir is effective for the treatment of CMV retinitis and is used experimentally to treat adenovirus, human papillomavirus, BK polyomavirus, vaccinia, and poxvirus infections. Intravenous cidofovir must be administered with high-dose probenecid (2 g at 3 hours before the infusion and 1 g at 2 and 8 hours after), which blocks active tubular secretion and decreases nephrotoxicity. Before each infusion, cidofovir dosage must be adjusted for alterations in the calculated creatinine clearance or for the presence of urine protein, and aggressive adjunctive hydration is required. Initiation of cidofovir therapy is contraindicated in patients with existing renal insufficiency. Direct intravitreal administration of cidofovir is not recommended because of ocular toxicity.
The primary adverse effect of intravenous cidofovir is a dose-dependent proximal tubular nephrotoxicity, which may be reduced with prehydration using normal saline. Proteinuria, azotemia, metabolic acidosis, and Fanconi’s syndrome may occur. Concurrent administration of other potentially nephrotoxic agents (eg, amphotericin B, aminoglycosides, nonsteroidal anti-inflammatory drugs, pentamidine, foscarnet) should be avoided. Prior administration of foscarnet may increase the risk of nephrotoxicity. Other potential adverse effects include uveitis, ocular hypotony, and neutropenia (15–24%). Concurrent probenecid use may result in other toxicities or drug-drug interactions (see Chapter 36). Cidofovir is mutagenic, gonadotoxic, and embryotoxic, and causes hypospermia and mammary adenocarcinomas in animals.
 ANTIRETROVIRAL AGENTS
ANTIRETROVIRAL AGENTS
Substantial advances have been made in antiretroviral therapy since the introduction of the first agent, zidovudine, in 1987. Six classes of antiretroviral agents are currently available for use: nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors, CCR5 co-receptor antagonists (also called entry inhibitors), and HIV integrase strand transfer inhibitors (INSTIs) (Table 49–3). These agents inhibit HIV replication at different parts of the cycle (Figure 49–3). Structures of some of these drugs are shown in eFigure 49–3.1.
TABLE 49–3 Currently available antiretroviral agents.
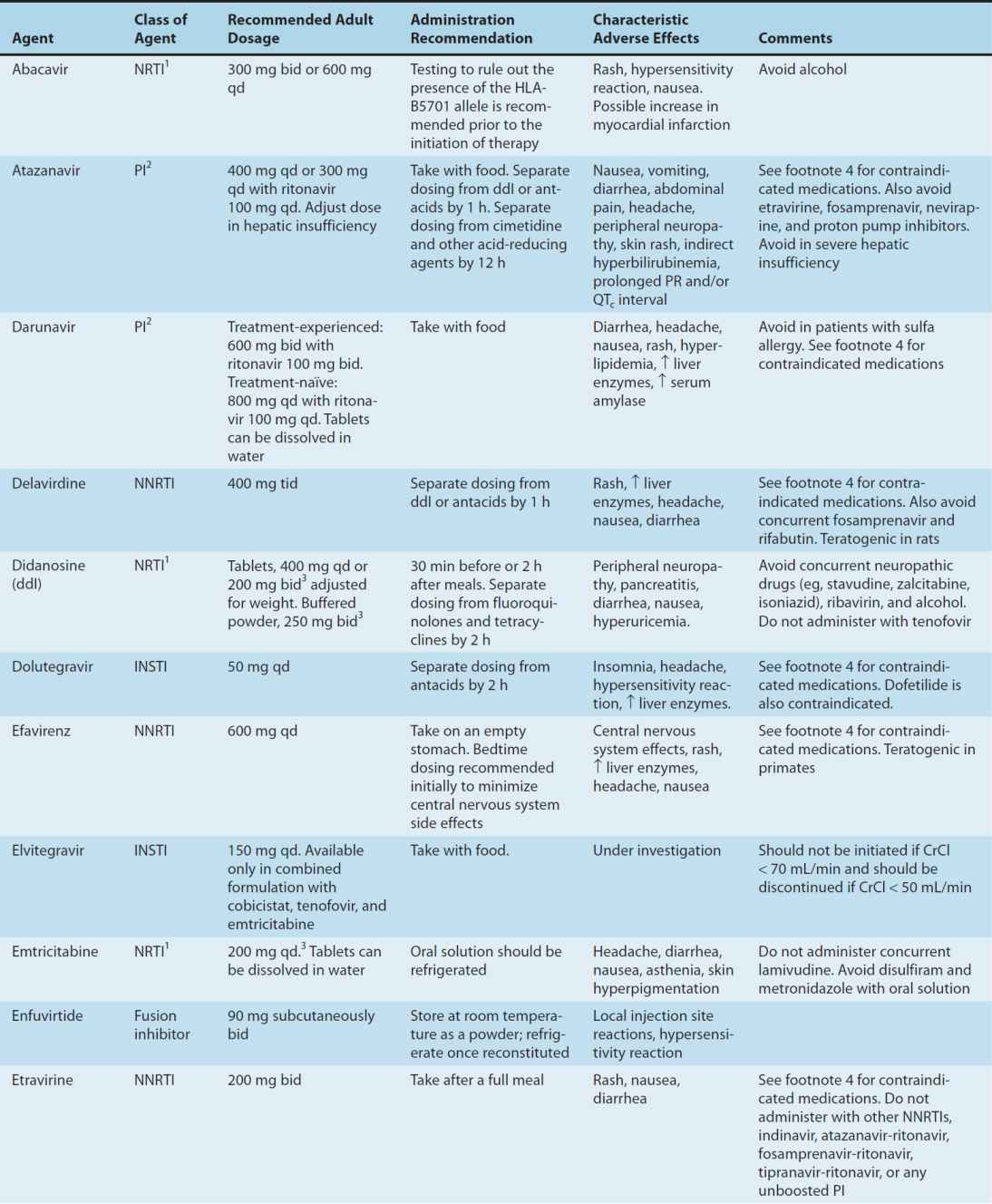
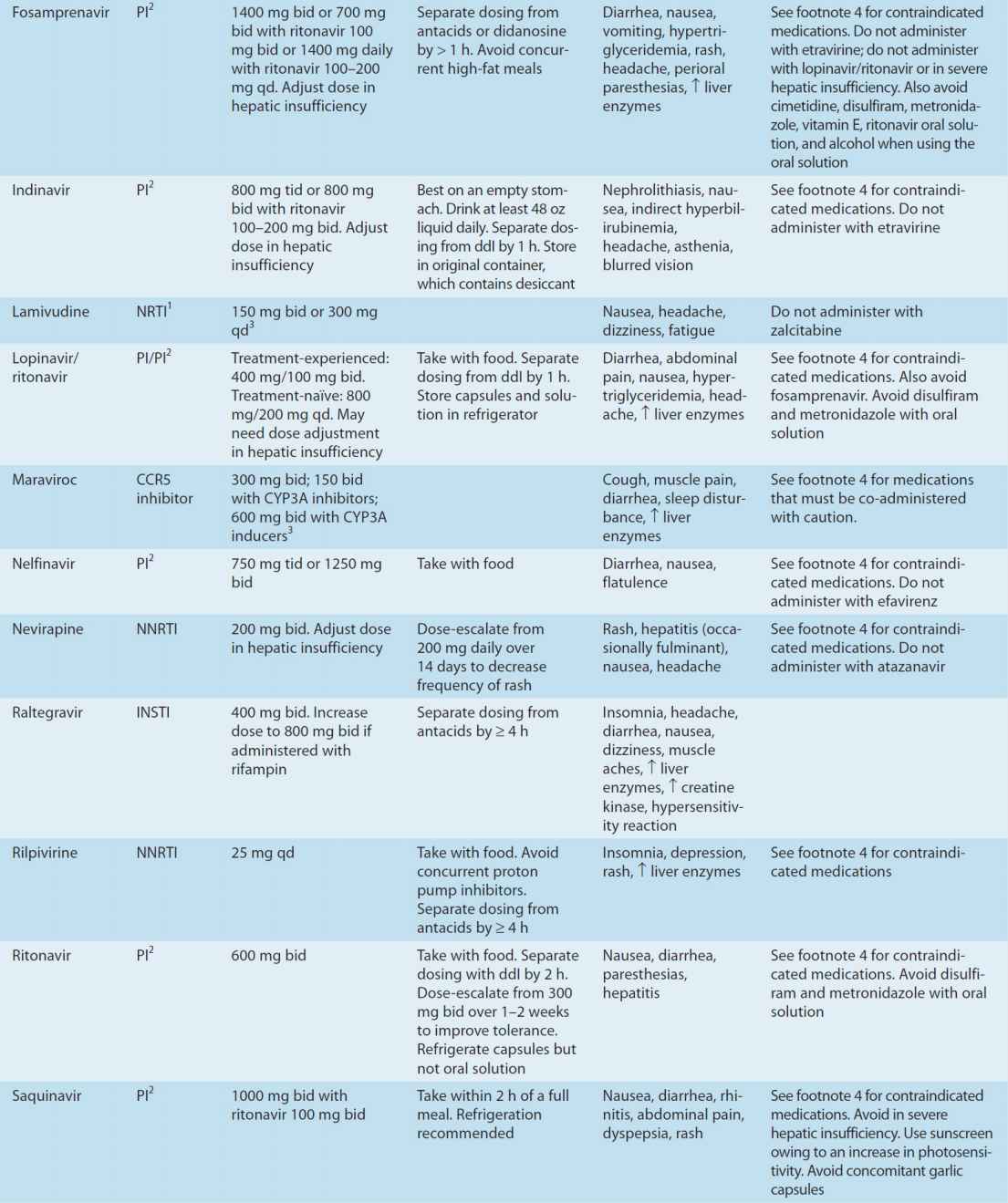
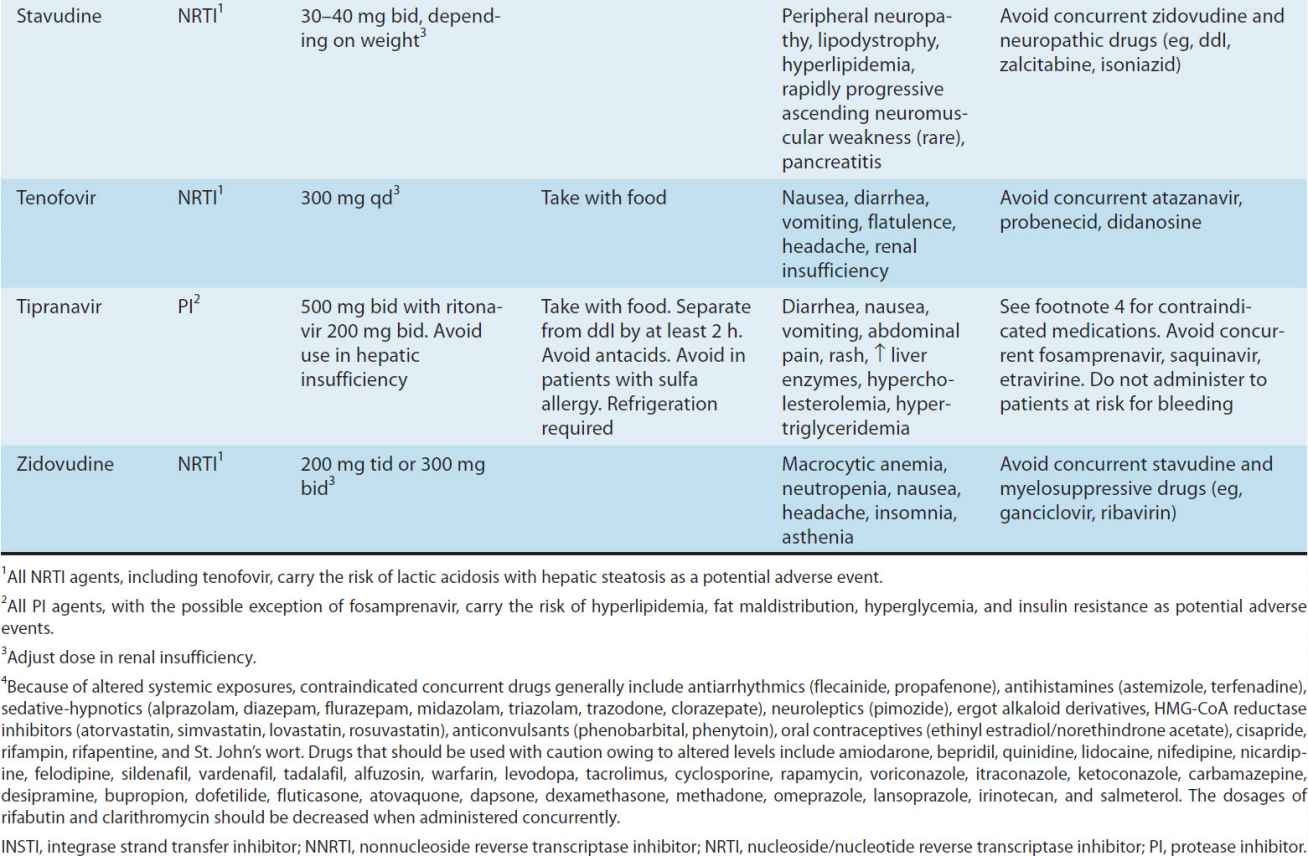
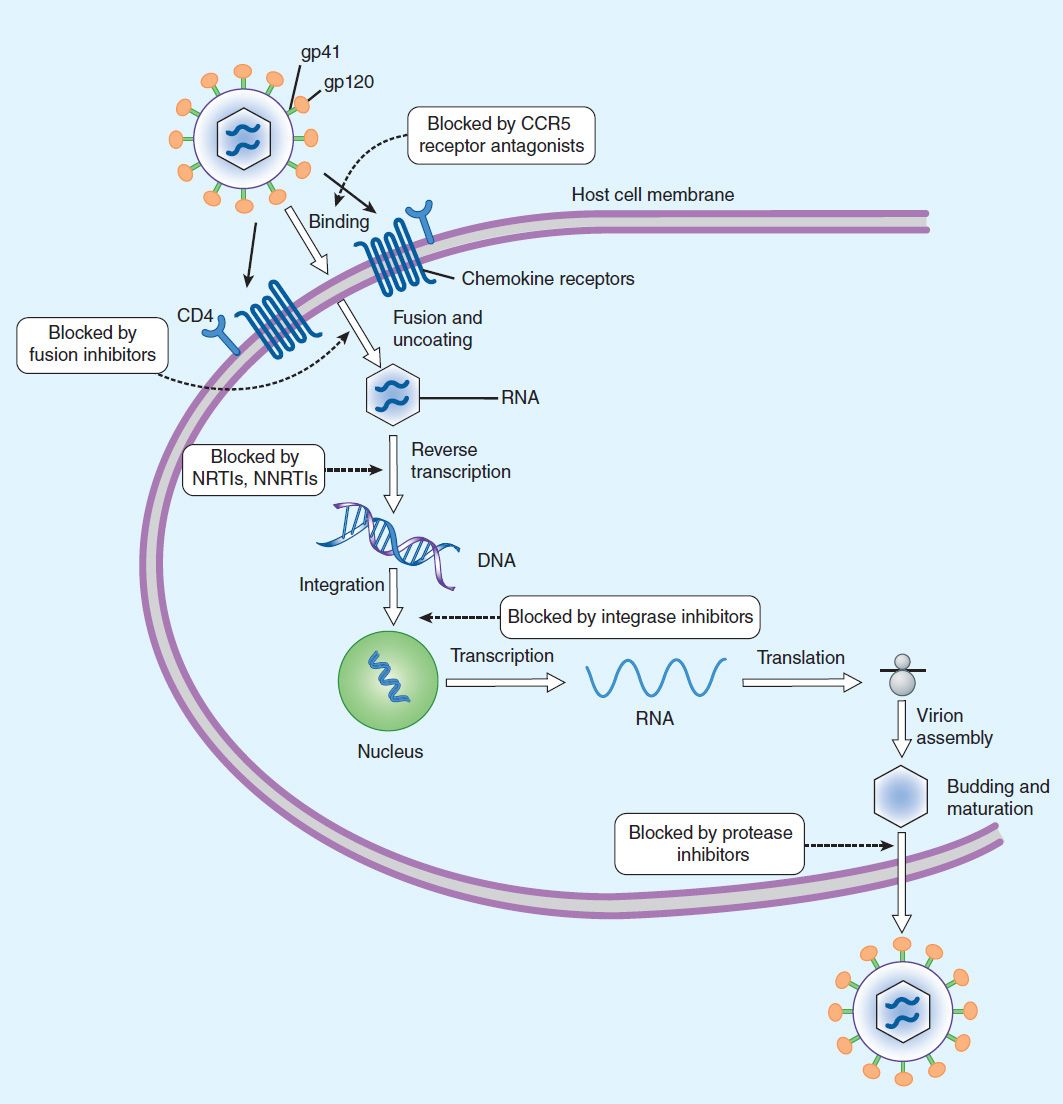
FIGURE 49–3 Life cycle of HIV. Binding of viral glycoproteins to host cell CD4 and chemokine receptors leads to fusion of the viral and host cell membranes via gp41 and entry of the virion into the cell. After uncoating, reverse transcription copies the single-stranded HIV RNA genome into double-stranded DNA, which is integrated into the host cell genome. Gene transcription by host cell enzymes produces messenger RNA, which is translated into proteins that assemble into immature noninfectious virions that bud from the host cell membrane. Maturation into fully infectious virions is through proteolytic cleavage. NNRTIs, nonnucleoside reverse transcriptase inhibitors; NRTIs, nucleoside/nucleotide reverse transcriptase inhibitors.
Greater knowledge of viral dynamics through the use of viral load and resistance testing has made it clear that combination therapy with maximally potent agents will reduce viral replication to the lowest possible level, thereby reducing the number of cumulative mutations and decreasing the likelihood of emergence of resistance. Thus, administration of combination antiretroviral therapy, typically including at least three antiretroviral agents with differing susceptibility patterns, has become the standard of care. Viral susceptibility to specific agents varies among patients and may change with time. Therefore, such combinations must be chosen with care and tailored to the individual, as must changes to a given regimen. In addition to potency and susceptibility, important factors in the selection of agents for any given patient are tolerability, convenience, and optimization of adherence. As new agents have become available, several older ones have had diminished usage, because of either suboptimal safety or inferior antiviral efficacy. Zalcitabine (ddC; dideoxycytidine), for example, is no longer marketed.
Decrease of the circulating viral load by antiretroviral therapy is correlated with enhanced survival as well as decreased morbidity. Also, recent evidence suggests that in addition to providing clinical benefits for the patient, the use of antiretroviral therapy strongly reduces the risk for heterosexual HIV transmission.
Discussion of antiretroviral agents in this chapter is specific to HIV-1. Patterns of susceptibility of HIV-2 to these agents may vary; however, there is generally innate resistance to the NNRTIs and lower barriers of resistance to NRTIs and PIs; data regarding maraviroc are inconclusive.
NUCLEOSIDE & NUCLEOTIDE REVERSE TRANSCRIPTASE INHIBITORS (NRTIs)
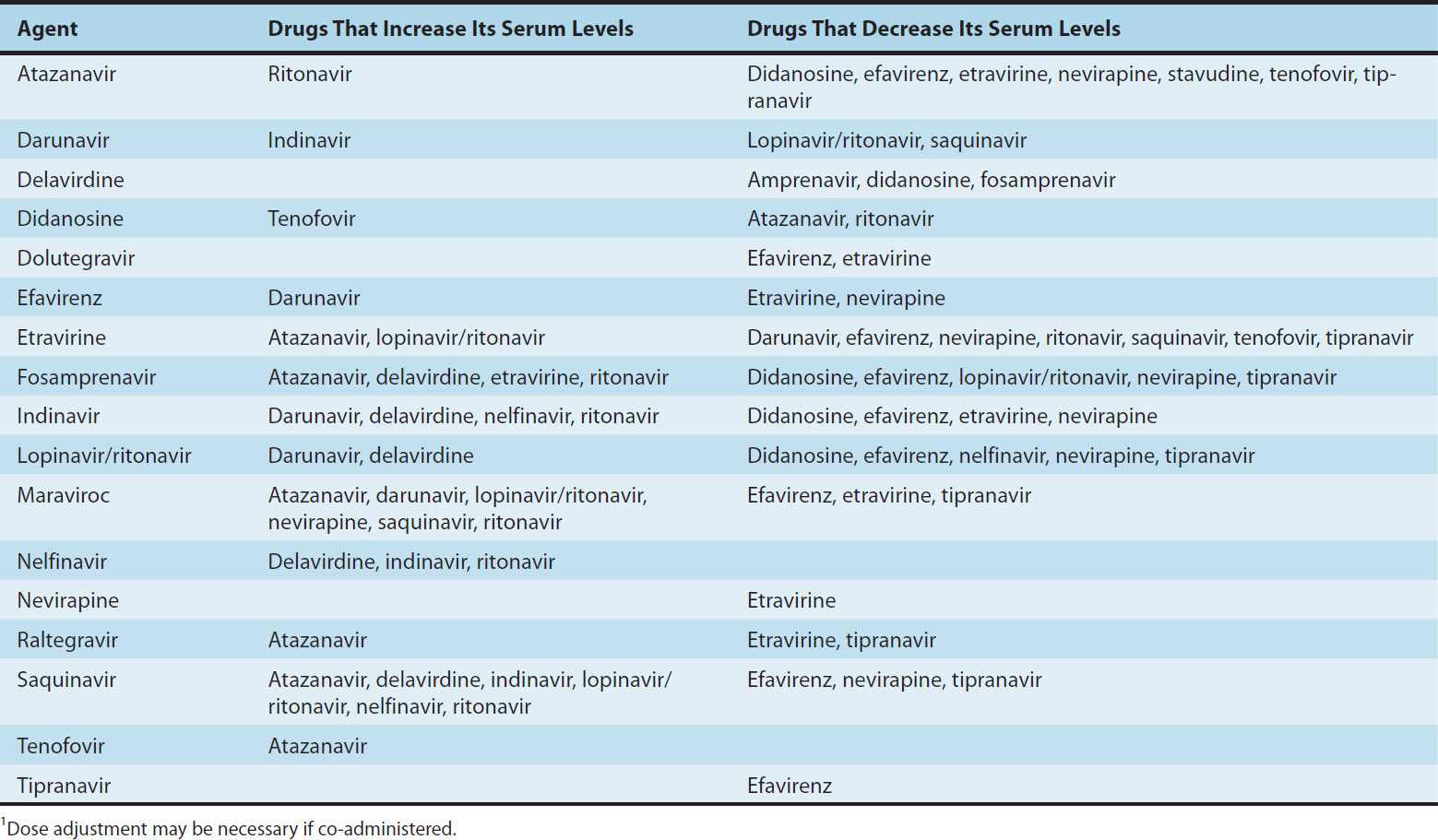
NRTIs are considered the “backbone” of antiretroviral therapy and are generally used in combination with other classes of agents, such as an NNRTI, PI, or integrase inhibitor. NRTIs are usually given in pairs, and many are available as coformulations in order to decrease pill burden and improve adherence. However, certain NRTI combinations should be avoided, due to either drug-drug interactions (eg, didanosine plus tenofovir; see Table 49–4), similar resistance patterns (eg, lamivudine plus emtricitabine) or overlapping toxicities (eg, stavudine plus didanosine).
TABLE 49–4 Clinically significant drug-drug interactions pertaining to two-drug antiretroviral combinations.1
The NRTIs act by competitive inhibition of HIV-1 reverse transcriptase; incorporation into the growing viral DNA chain causes premature chain termination due to inhibition of binding with the incoming nucleotide (Figure 49–3). Each agent requires intracytoplasmic activation via phosphorylation by cellular enzymes to the triphosphate form.
Typical resistance mutations include M184V, L74V, D67N, and M41L. Lamivudine or emtricitabine therapy tends to select rapidly for the M184V mutation in regimens that are not fully suppressive. While the M184V mutation confers reduced susceptibility to abacavir, didanosine, and zalcitabine, its presence may restore phenotypic susceptibility to zidovudine. The K65R/N mutation is associated with reduced susceptibility to tenofovir, abacavir, lamivudine, and emtricitabine.
All NRTIs may be associated with mitochondrial toxicity, probably owing to inhibition of mitochondrial DNA polymerase gamma. Less commonly, lactic acidosis with hepatic steatosis may occur, which can be fatal. NRTI treatment should be suspended in the setting of rapidly rising aminotransferase levels, progressive hepatomegaly, or metabolic acidosis of unknown cause. The thymidine analogs zidovudine and stavudine may be particularly associated with dyslipidemia and insulin resistance. Also, some evidence suggests an increased risk of myocardial infarction in patients receiving abacavir; this remains unproven.
ABACAVIR
Abacavir is a guanosine analog that is well absorbed following oral administration (83%) and is unaffected by food. The serum half-life is 1.5 hours. The drug undergoes hepatic glucuronidation and carboxylation. Since the drug is metabolized by alcohol dehydrogenase, serum levels of abacavir may be increased with concurrent alcohol (ie, ethanol) ingestion. Cerebrospinal fluid levels are approximately one-third those of plasma. Abacavir is available in a fixed dose formulation with lamivudine and also with zidovudine plus lamivudine.
High-level resistance to abacavir appears to require at least two or three concomitant mutations and thus tends to develop slowly.
Hypersensitivity reactions, occasionally fatal, have been reported in up to 8% of patients receiving abacavir and may be more severe in association with once-daily dosing. Symptoms, which generally occur within the first 6 weeks of therapy, include fever, fatigue, nausea, vomiting, diarrhea, and abdominal pain. Respiratory symptoms such as dyspnea, pharyngitis, and cough may also be present, and skin rash occurs in about 50% of patients. The laboratory abnormalities of a mildly elevated serum aminotransferase or creatine kinase level may be present but are nonspecific. Although the syndrome tends to resolve quickly with discontinuation of medication, rechallenge with abacavir results in return of symptoms within hours and may be fatal. Screening for HLA-B*5701 before initiation of abacavir therapy is recommended to identify patients with a markedly increased risk for abacavir-associated hypersensitivity reaction. Although the positive predictive value of this test is only about 50%, it has a negative predictive value approaching 100%.
Other potential adverse events are rash, fever, nausea, vomiting, diarrhea, headache, dyspnea, fatigue, and pancreatitis (rare). In some studies but not in others, abacavir has been associated with a higher risk of myocardial infarction. Since abacavir may lower methadone levels, patients receiving these two agents concurrently should be monitored for signs of opioid withdrawal and may require an increased dose of methadone.
DIDANOSINE
Didanosine (ddI) is a synthetic analog of deoxyadenosine. Oral bioavailability is approximately 40%. Dosing on an empty stomach is optimal, but buffered formulations are necessary to prevent inactivation by gastric acid (Table 49–3). Cerebrospinal fluid concentrations of the drug are approximately 20% of serum concentrations. Serum half-life is 1.5 hours, but the intracellular half-life of the activated compound is as long as 20–24 hours. The drug is eliminated by both cellular metabolism and renal excretion.
The major clinical toxicity associated with didanosine therapy is dose-dependent pancreatitis. Other risk factors for pancreatitis (eg, alcohol abuse, hypertriglyceridemia) are relative contraindications, and concurrent use of drugs with the potential to cause pancreatitis, including zalcitabine, stavudine, ribavirin, and hydroxyurea, should be avoided (Table 49–3). The risk of peripheral distal sensory neuropathy, another potential toxicity, may be increased with concurrent use of stavudine, isoniazid, vincristine, or ribavirin. Other reported adverse effects include diarrhea (particularly with the buffered formulation), hepatitis, esophageal ulceration, cardiomyopathy, central nervous system toxicity (headache, irritability, insomnia), and hypertriglyceridemia. Due to an increased risk of lactic acidosis and hepatic steatosis when combined with stavudine, this combination should be avoided, especially during pregnancy. Previously asymptomatic hyperuricemia may precipitate attacks of gout in susceptible individuals; concurrent use of allopurinol may increase levels of didanosine. Reports of retinal changes and optic neuritis in patients receiving didanosine, particularly in adults receiving high doses and in children, mandate periodic retinal examinations. Lipoatrophy appears to be more common in patients receiving didanosine or other thymidine analogs.
The buffer in didanosine tablets interferes with absorption of indinavir, delavirdine, atazanavir, dapsone, itraconazole, and fluoroquinolone agents; therefore, administration should be separated in time. Serum levels of didanosine are increased when co-administered with tenofovir or ganciclovir, and are decreased by atazanavir, delavirdine, ritonavir, tipranavir, and methadone (Table 49–4). Didanosine should not be used in combination with ribavirin.
EMTRICITABINE
Emtricitabine (FTC) is a fluorinated analog of lamivudine with a long intracellular half-life (> 24 hours), allowing for once-daily dosing. Oral bioavailability of the capsules is 93% and is unaffected by food, but penetration into the cerebrospinal fluid is low. Elimination is by both glomerular filtration and active tubular secretion. The serum half-life is about 10 hours.
The oral solution, which contains propylene glycol, is contraindicated in young children, pregnant women, patients with renal or hepatic failure, and those using metronidazole or disulfiram. Also, because of its activity against HBV, patients co-infected with HIV and HBV should be closely monitored if treatment with emtricitabine is interrupted or discontinued, owing to the likelihood of hepatitis flare.
Emtricitabine is available in a fixed-dose formulation with tenofovir, either alone or in combination with efavirenz, rilpivirine, or elvitegravir plus cobicistat (a boosting agent). Based on results of clinical trials, the combination of tenofovir and emtricitabine is now recommended as pre-exposure prophylaxis to reduce HIV acquisition in men who have sex with men, in heterosexually active men and women, and in injection drug users.
Like lamivudine, the M184V/I mutation is most frequently associated with emtricitabine use and may emerge rapidly in patients receiving regimens that are not fully suppressive. Because of their similar mechanisms of action and resistance profiles, the combination of lamivudine and emtricitabine is not recommended.
The most common adverse effects observed in patients receiving emtricitabine are headache, insomnia, nausea, and rash. In addition, hyperpigmentation of the palms or soles may be observed (~ 3%), particularly in African-Americans (up to 13%).
LAMIVUDINE
Lamivudine (3TC) is a cytosine analog (eFigure 49–3.1) with in vitro activity against HIV-1 that is synergistic with a variety of antiretroviral nucleoside analogs—including zidovudine and stavudine—against both zidovudine-sensitive and zidovudine-resistant HIV-1 strains. As with emtricitabine, lamivudine has activity against HBV; therefore, discontinuation in patients that are co-infected with HIV and HBV may be associated with a flare of hepatitis. Lamivudine therapy rapidly selects for the M184V mutation in regimens that are not fully suppressive.
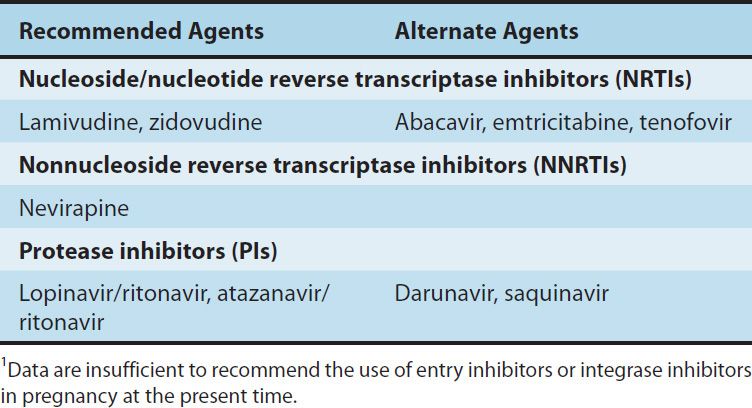
Oral bioavailability exceeds 80% and is not food-dependent. In children, the average cerebrospinal fluid:plasma ratio of lamivudine was 0.2. Serum half-life is 2.5 hours, whereas the intracellular half-life of the triphosphorylated compound is 11–14 hours. Most of the drug is eliminated unchanged in the urine. Lamivudine remains one of the recommended antiretroviral agents in pregnant women (Table 49–5). Lamivudine is available in a fixed-dose formulation with zidovudine and also with abacavir.
TABLE 49–5 The use of antiretroviral agents in pregnancy.1
Potential adverse effects are headache, dizziness, insomnia, fatigue, dry mouth, and gastrointestinal discomfort, although these are typically mild and infrequent. Lamivudine’s bioavailability increases when it is co-administered with trimethoprim-sulfamethoxazole. Lamivudine and zalcitabine may inhibit the intracellular phosphorylation of one another; therefore, their concurrent use should be avoided if possible.
STAVUDINE
The thymidine analog stavudine (d4T) has high oral bioavailability (86%) that is not food-dependent. The serum half-life is 1.1 hours, the intracellular half-life is 3.0–3.5 hours, and mean cerebrospinal fluid concentrations are 55% of those of plasma. Excretion is by active tubular secretion and glomerular filtration.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


