Antiviral Agents
Donald M. Coen
Douglas D. Richman
Overview
Medical Importance of Antiviral Drugs and Barriers to Their Development
Viruses are a leading cause of disease and death worldwide. Although public health measures and vaccines are the most effective ways to control many viral infections, preventive measures have not succeeded for numerous viral diseases. For some of these diseases, antiviral drugs have been developed. A number of these drugs have been highly successful, saving lives and relieving suffering. This has been most dramatic with drugs that are active against human immunodeficiency virus (HIV). In the developed world, these drugs have transformed a progressive fatal disease into a manageable condition, in which virus replication can be suppressed presumably for a lifetime as long as antiretroviral therapy is maintained. These dramatic benefits had been extended to more than 7 million people globally by 2011.182
Despite these successes, there are still relatively few diseases for which highly effective antiviral drugs have been developed. There are several factors that contribute to this. With very few exceptions, such as the polymerases of related viruses, drug targets are virus specific, and therefore only a few antiviral drugs developed against one virus have been active against other viruses. Some viruses (e.g., herpesviruses, HIV) establish latent infections, so treating active infections does not cure the patient. Different viruses, especially respiratory viruses, can cause similar symptoms, making diagnosis difficult. Often, treatment must begin early to be beneficial, sometimes because the viral diseases are quickly controlled by immune responses.
On top of these factors are economic issues that have often discouraged pharmaceutical companies from developing antiviral drugs. For example, any individual virus disease may represent a relatively small potential market. It is not surprising then that companies have focused mainly on easily diagnosed and chronic viral diseases that require prolonged drug administration and that are relatively widespread in developed countries, because these are diseases for which treatment would earn substantial profits. Perhaps ironically, these diseases are often caused by viruses that establish latent infections and are not curable.
The first highly successful antiviral drug was acyclovir (ACV), initially developed in the late 1970s against herpes simplex virus (HSV) 1 and 2 and varicella zoster virus (VZV).91 Antiviral drug discovery expanded markedly with the acquired immunodeficiency syndrome (AIDS) epidemic, which led to a variety of drugs against HIV and opportunistic pathogens such as human cytomegalovirus (HCMV). This coincided with expanding knowledge about virus genetics, molecular biology, enzymology, and protein structure, which permitted modern, more rational approaches, to drug discovery. Indeed, antiviral drugs have been a proving ground for these approaches.
The Process of Antiviral Drug Development
Older antiviral drugs such as amantadine and ACV were discovered by the testing of chemicals for their ability to inhibit virus replication in labor- and time-intensive assays of viral replication, such as assays of cytopathic effect or plaque assays.75,327 These chemicals had typically been synthesized for some other
purpose, and discovery of their antiviral activities was often serendipitous. When particular chemicals were known to have antiviral activity, a variety of other chemicals with similar structures were tested for activity. Several examples of this somewhat more rational procedure can be found in the sections on specific drugs.
purpose, and discovery of their antiviral activities was often serendipitous. When particular chemicals were known to have antiviral activity, a variety of other chemicals with similar structures were tested for activity. Several examples of this somewhat more rational procedure can be found in the sections on specific drugs.
The more modern and rational approach of high-throughput screening of large libraries of chemicals has been used to discover some newer drugs. High throughput screens can be “target-based” or “cell-based.” Target-based screens start with the identification of a viral gene product that would make a good drug target (see below for discussion). It is important that the target protein can be expressed at high levels and purified, and that it can be assayed rapidly with high-throughput robotic technology. Such target-based screens can examine more than one million chemical compounds in no more than a few weeks. Most nonnucleoside reverse transcriptase inhibitors (NNRTIs), which are active against HIV-1, were discovered using this approach (e.g., nevirapine246). Cell-based high-throughput screens are slower, more cumbersome, and more expensive. However, such assays permit interrogation of multiple and unappreciated mechanisms of action, and can be performed relatively rapidly using robots (i.e., within one to a few days per assay). In addition, they automatically identify compounds that can enter cells and block a step of virus replication. Highly potent compounds that target a poorly understood function of hepatitis C virus (HCV) and block HCV replication in vivo have been discovered using a cell-based screen.110
A still more rational approach is to exploit detailed knowledge about a viral protein to design drugs that will inhibit its activity. In particular, knowledge of the three-dimensional structure of the protein can enable visualization of its active site and understanding of its mechanism of action, and can also be used to design or refine small molecules that can block the active site and the protein’s action. An example of a drug discovered using this approach is the neuraminidase inhibitor, zanamivir, which is active against influenza A and B viruses.370
The approaches outlined in the preceding text result in what is called a “hit”; however, a hit is only an inhibitor of the function of a viral protein or of viral replication in cell culture. The next steps involve using medicinal chemistry to convert the hit into a compound with high potency and desirable pharmacological properties (e.g., bioavailability, long elimination half-life, low toxicity), not to mention patentability. If the structure of the hit compound bound to its target is available, this information can be used to help generate additional compounds to be tested. This process will be illustrated below with the example of the anti-HIV protease inhibitor, ritonavir. Simultaneously, studies to demonstrate the mechanism(s) of action of the compounds are undertaken (see section on antiviral drug mechanisms and drug resistance below). A key, but often overlooked aspect of drug development is developing optimal methods for synthesis of pure compound on a large scale and formulation of the compound for administration to humans.
When a candidate compound with desirable properties has been developed, clinical trials begin. There is no rigid formula by which an antiviral candidate is assessed after it enters the clinic. Typically, the process of clinical development starts with the administration of drug in escalating doses to small cohorts of volunteers to assess pharmacokinetics and tolerability (phase I trials). With some virus infections, these studies may be performed in patients in whom drug activity can also be assessed, for example by quantifying reductions of chronic HCMV shedding in the genitourinary tract or of HCV in the plasma (proof-of-concept studies). Substantial reductions in viral load encourage progression to more advanced clinical trials. However, with an infection such as HIV, the risk of selecting for drug resistance that might compromise future treatment options often restricts phase I trials to studies of HIV-seronegative subjects. This issue may become important for other viruses (e.g., HCV).
A major advantage of antiviral drug development, compared to drug development for many other diseases, is that quantitative reductions in virus shedding are a precise measure of activity and in general correlate with clinical end points. These quantitative reductions can be measured quickly (days) and permit determination of whether drugs work and of appropriate doses and dosing intervals. Phase II clinical trials assess this antiviral activity along with additional pharmacological and safety information in dozens to several hundred subjects. The data from phase II trials permit the design of larger phase III registrational studies, which provide statistically robust results that fulfill regulatory requirements for drug approval. Trials for drugs for influenza and herpesviruses infections require clinical end points (i.e., amelioration of disease), with virological data supporting those. For HIV, and increasingly for hepatitis B virus (HBV) and HCV, the correlations between levels of viremia and disease, and the correlations between drug-induced reductions in viremia and improvement of disease have resulted in virological end points (quantitative reduction and proportion of patients below the limit of detection) as criteria for drug approval.
Phase III studies of a new antiretroviral drug can pursue two general indications. For patients who failed prior regimens because of drug-resistant virus and who have limited treatment options, an accelerated approval process is available with shorter trials to document efficacy. Historically, patients were randomized to an optimized regimen based on the results of a drug resistance test with or without the addition of the test drug that presumably has activity against drug-resistant virus. Demonstration of superior virological end points after 24 weeks with continued follow-up for at least a year was considered supportive of an unmet medical need to fulfill criteria for registrational approval. Repeated demonstrations that this approach worked have led to the sense that placebo arms were no longer ethically defensible. The proper design of future trials for these patients remains unclear.206
Development of a drug for first-line (initial) therapy faces higher hurdles. Patients starting antiretroviral therapy are now usually asymptomatic and must plan for decades of successful suppression of virus replication. Therefore, a new drug must offer superiority to the current standard of care, and a combination including a new drug “X” (for example, tenofovir and emtricitabine [FTC] plus drug X) might be compared to tenofovir and FTC plus efavirenz. Hundreds of subjects in each arm must be followed for at least 2 years for efficacy and safety to fulfill regulatory criteria for approval of a drug for use in initial treatment. A predefined statistical definition of “noninferiority” is the criterion for approval of candidate drugs for which there is an approved drug in the same class,
Should the clinical trials yield the desired results, the “hit” will have turned into an approved drug. Even then, its efficacy and safety will be put to the test in much larger populations, and new indications for its use may emerge.
Should the clinical trials yield the desired results, the “hit” will have turned into an approved drug. Even then, its efficacy and safety will be put to the test in much larger populations, and new indications for its use may emerge.
Importance of Antiviral Drugs for Basic Science
Aside from their importance in treating viral diseases, antiviral drugs make excellent laboratory tools. As is illustrated in detail later in this chapter, drugs can block the functions of specific viral proteins and thus specific stages in viral replication. This is crucial for understanding the timing of particular events in the virus replication cycle and which proteins are needed for each step. Studies using selective antiviral drugs, coupled with viral mutations that confer drug resistance, can help to identify the roles of viral proteins and dissect the details of how these proteins function. Drug-resistance mutations can provide selectable markers for engineering interesting mutant viruses and performing viral genetics.
Examples of how studies of antiviral drugs and drug resistance can illuminate virus biology and biochemistry include the role of amantadine in the discovery of an influenza A ion channel and its function in virus uncoating,142,289 and the role of ganciclovir in the discovery of an unusual HCMV protein kinase that can phosphorylate nucleoside analogs.350 Crystal structures of drug targets with bound inhibitors have yielded important insights into structure and function of these targets. For example, the first structure of a retroviral (HIV) reverse transcriptase was obtained in complex with nevirapine.193 Similarly, clinical investigation has been greatly abetted by antiviral drugs. Intensive monitoring of the dynamics of HIV replication following the administration of drug has permitted important insights about rates of virus production, virus clearance, and cellular sources of virus replication.129,282 Studies of patients receiving HIV therapy have elucidated much of our understanding of HIV latency, evolution, and fitness.50,98,390 Finally, the remarkable restoration of CD4 cell numbers and function with HIV therapy provides insights regarding both lymphocyte dynamics and immune function.7,129
General Aspects of Antiviral Drug Mechanisms and Drug Resistance
Drug Selectivity, Resistance, and Drug Targets
The value of antiviral drugs to basic science depends on how selective they are and on how well their mechanisms are understood. Selectivity and mechanism are also crucial for the clinical use of antiviral drugs. Biologically, selectivity is the difference between the dose of the drug that exerts its antiviral effect (usually quantified as the dose that reduces viral activity by 50%) and the dose that exerts its cytotoxic effect (or more subtle impairments of the host). Examples of how selectivity is achieved biochemically can be found among the descriptions of the mechanisms of specific antiviral drugs.
It is generally agreed that the best way to understand antiviral selectivity and mechanism is through the study of drug resistance. Because viruses are obligate intracellular parasites, the detection of resistance to an antiviral drug implies that the drug is selective. That is, the drug acts at least in part by interfering directly with a virus-specific process, rather than by incapacitating the host cell.157 In much the same way, a mutation in a viral gene that confers resistance to an antiviral drug implies that the gene or its product is necessary for at least part of the selective action of the drug. It is not enough to identify a mutation in a drug-resistant mutant to show that the mutation confers resistance. It is crucial to perform a genetic experiment (e.g., reconstruct a virus with the mutation [and no other] and show that the reconstructed virus is drug-resistant).
In most cases, drug-resistant mutations identify targets of antiviral drugs. However, not all drug-resistant mutations identify drug targets. For example, inhibitors of influenza virus neuraminidase (e.g., zanamivir) can select for mutations in the gene encoding hemagglutinin.125,241,341 These mutations would be expected to result in a hemagglutinin that binds less avidly to cell surface glycoproteins. This in turn could result in a reduced requirement for neuraminidase to release the virion from the cell, and thus resistance to the neuraminidase inhibitor. Accordingly, genetics alone is not sufficient to identify a drug target. Ideally, one should also show that the drug interacts with the target, and that the resistance mutation results in a target with an altered interaction with the drug that explains the resistance phenotype. This usually requires biochemical studies, and the analyses are not always straightforward.
Genetics alone is also not sufficient to reveal the detailed mechanism of action of an inhibitor. Mapping a resistance mutation that results in a single amino acid change in the target protein identifies that residue as being necessary for sensitivity to the drug. Other genetic approaches, such as the generation of chimeric genes from closely related viruses that are susceptible or resistant to a drug (e.g., HIV-1 and HIV-2, and NNRTIs67), can identify residues that are sufficient to confer susceptibility to the drug to an otherwise resistant protein. However, these results do not identify residues that interact with the drug. Identification of a drug-binding site requires methods such as photoaffinity cross-linking and x-ray crystallography. These two methods have identified the binding site for the NNRTI, nevirapine, on HIV-1 reverse transcriptase (RT).62,193,392 Mapping a drug binding site is valuable, but does not tell us how drug binding exerts its physiologic action. In the case of NNRTIs, pre–steady-state kinetic analyses of HIV-1 RT were used to show that the drug slows the enzyme’s incorporation of deoxynucleoside triphosphates (dNTPs) into DNA,313,338 presumably by altering the orientation of catalytic residues, as suggested by the location of the drug binding site.193 Therefore, solving the mechanism of action of an antiviral drug requires multidisciplinary approaches.
For a number of antiviral drugs, we still lack a complete understanding of their mechanisms of action and resistance, and, in some cases, there is considerable controversy regarding these mechanisms. This can even be true for drugs that are designed against a specific target. For example, fomivirsen is an oligonucleotide drug that is designed to inhibit HCMV replication by its complementarity to messenger RNA (mRNA) for an important regulatory protein.3,8 However, studies of a drug-resistant mutant did not confirm this mechanism of action.262 Therefore, the mechanism of fomivirsen remains unknown. We will not discuss this and certain other drugs such as the anti-HSV topical treatment, docosanol, whose mechanisms are unknown.
General Aspects of Antiviral Drug Targets
There are two major kinds of virus proteins that serve as antiviral drug targets. In general, it is easier to inhibit a protein’s
function (with an antagonist) than to promote its function (with an agonist). Therefore, the most common antiviral drug target is a virus-encoded protein that can be inhibited by a drug. Ideally, this kind of target should be essential for viral replication. Less ideally, it should at least be very important for replication and pathogenesis in the human host. The target should also be sufficiently different from any important host proteins to permit selectivity. It should be druggable; that is, a small molecule should be able to inhibit it. Enzymes make especially good targets for inhibition because they are usually at low concentrations inside cells, they are relatively well-understood mechanistically, they often interact with small-molecule substrates, and, as catalysts, their inhibition interrupts multiple reactions. Furthermore, pharmaceutical companies have a great deal of experience in developing enzyme inhibitors as drugs. It is therefore not surprising that most antiviral drug targets are virus-encoded enzymes. A second kind of target is a virus-encoded protein that can activate the drug to make it inhibitory to the virus (lethal synthesis), and that is sufficiently different from host counterparts to permit selectivity. An example is HSV thymidine kinase (TK), which activates ACV and its congeners.108
function (with an antagonist) than to promote its function (with an agonist). Therefore, the most common antiviral drug target is a virus-encoded protein that can be inhibited by a drug. Ideally, this kind of target should be essential for viral replication. Less ideally, it should at least be very important for replication and pathogenesis in the human host. The target should also be sufficiently different from any important host proteins to permit selectivity. It should be druggable; that is, a small molecule should be able to inhibit it. Enzymes make especially good targets for inhibition because they are usually at low concentrations inside cells, they are relatively well-understood mechanistically, they often interact with small-molecule substrates, and, as catalysts, their inhibition interrupts multiple reactions. Furthermore, pharmaceutical companies have a great deal of experience in developing enzyme inhibitors as drugs. It is therefore not surprising that most antiviral drug targets are virus-encoded enzymes. A second kind of target is a virus-encoded protein that can activate the drug to make it inhibitory to the virus (lethal synthesis), and that is sufficiently different from host counterparts to permit selectivity. An example is HSV thymidine kinase (TK), which activates ACV and its congeners.108
Host proteins may also serve as antiviral drug targets, if they have an activity that is more important for viral replication than for host functions. A successful example of this approach is targeting the cell surface molecule, C-C chemokine receptor type 5 (CCR5), which led to the development of the anti-HIV drug maraviroc.207,297 Targeting of host functions is inherently more likely to result in toxicity than is targeting of viral functions. Conversely, it has been argued that viruses should be less likely to develop resistance against drugs that target host functions. Regardless, such mutants certainly can develop (e.g.,328,366).
Factors Affecting the Development of Drug Resistance
If a drug acts selectively against a virus, the virus will generally be able to develop resistance to it. However, the frequency of drug-resistant mutants in a population, and how rapidly they arise depend on several factors: The first is the mutation rate of the virus. The higher the mutation rate, the more rapidly resistance can develop. Viral mutation rates are largely controlled by the fidelity of the polymerases that replicate the viral genomes. RNA viruses, which replicate using low-fidelity RNA polymerases, usually have the highest mutation rates, averaging one mutation per genome per replication cycle,87,232 whereas DNA viruses, whose DNA polymerases include proofreading 3′ to 5′ exonucleases that contribute to fidelity,172 have lower mutation rates. However, other factors, including nucleotide pools, which can be modulated by both viral and cellular factors, also contribute to mutation rate.
The second factor is the target size for mutation. The more sites where mutations can confer drug resistance, the more rapidly resistance can arise. For some drugs, such as ACV, many mutations can confer resistance, as any mutation that substantially reduces viral TK activity results in resistance. In addition, some sites within a gene are more or less likely to mutate than others. Homopolymeric runs in the HSV tk gene (e.g., a run of seven G’s) are hot spots for frameshift mutations that confer ACV resistance.170
The third factor is magnitude of replication of the virus. The more copies of viral genomes produced, the more opportunities for resistance to arise. The fourth factor is the preexisting size of the virus population. The more virus present, even in the absence of drug selection, the more likely that drug-resistant mutants will be present among the array of genetic variants. Examples of these two factors that have an impact on HIV drug resistance are provided below in the section on drug resistance in antiviral therapy.
The fifth factor involves fitness (i.e., how well a genetic variant reproduces relative to other genetic variants, which can include “wild-type”). The more fit, the more likely resistance will occur. This is especially important for clinically significant drug resistance. For a virus to cause disease that is resistant to an antiviral drug, it must mutate not only to evade drug action, but also to retain pathogenicity. In vivo fitness and pathogenicity can be inferred from clinical studies, but can be assessed more directly in animal models. Although there is always the question of whether animal models are predictive of human disease, in most cases, there has been an excellent correlation between how drug-resistant mutants behave in clinical and animal studies. Some drug-resistant mutants, such as influenza viruses that are resistant to adamantanes, can be highly fit, both in cell culture and in animal models.15,355 Moreover, such mutants appear to be highly fit in human infections as well (reviewed in 144). Most ACV-resistant HSV mutants are highly fit in cell culture, but are much less so in animal models, which correlates with the low frequency that these mutants are recovered in immunocompetent humans (reviewed in 58 and 199). Still other drug-resistant mutants, such as certain HCMV mutants that are resistant to the drug foscarnet, can be less fit in cell culture, but not so impaired that they do not cause disease in patients.12 Relative fitness is important both in the absence and in the presence of drug. If wild-type virus can replicate relatively well in the presence of drug (e.g., at subtherapeutic concentrations), then resistant mutants will be less likely to predominate. These various factors—mutation rate, target size, replication rate, pre-existing population, and fitness—can be taken into account in mathematical formulas and used to model the emergence of drug resistance (e.g.61).
In addition, these factors influence how different viral infections are treated. For example, HIV infections are now treated with combinations of antiviral drugs, whereas HSV infections are treated with single agents. This is discussed later in the sections on antiviral therapy.
Mechanisms of Specific Antiviral Drugs
This section mainly considers agents that are currently approved for clinical use. No endorsement of the therapeutic value of these drugs is intended. For certain viruses that can be studied usefully in animal models, we discuss the effects of drug-resistant mutations on replication and pathogenesis in vivo.
Targeting Drugs to Specific Stages of Virus Infection
As detailed throughout this book, the infection of cells by viruses can be broken down into a common set of steps or stages, and most of these stages have been targeted by approved antiviral drugs (Fig. 13.1). The order of stages can differ for different viruses, and some viruses have stages that are not
shared with other viruses. For example, the replication of HIV and the stages that have been targeted by approved antiviral drugs are illustrated in Figure 13.2. In principle, any stage can be targeted for inhibition. There are potential advantages to targeting very early or late stages such as attachment, entry, and release, because inhibitors of these stages do not have to enter cells to exert activity. Stages such as genome replication, assembly, and maturation often require specific viral enzymes, which, as described earlier, are attractive drug targets. Indeed, most antiviral drugs currently available inhibit genome replication. Nevertheless, there are antiviral drugs that act at nearly every stage of viral infection.
shared with other viruses. For example, the replication of HIV and the stages that have been targeted by approved antiviral drugs are illustrated in Figure 13.2. In principle, any stage can be targeted for inhibition. There are potential advantages to targeting very early or late stages such as attachment, entry, and release, because inhibitors of these stages do not have to enter cells to exert activity. Stages such as genome replication, assembly, and maturation often require specific viral enzymes, which, as described earlier, are attractive drug targets. Indeed, most antiviral drugs currently available inhibit genome replication. Nevertheless, there are antiviral drugs that act at nearly every stage of viral infection.
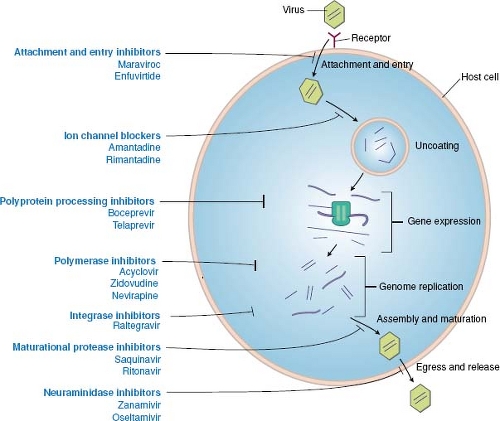 Figure 13.1. Antiviral drugs block various stages of viral replication. A generic replication cycle of viruses in cells is cartooned, showing the stages of infection, which different drug classes block. These include attachment and entry, uncoating, gene expression, genome replication, assembly and maturation, and egress and release. Examples of specific drug classes are provided. For some viruses (e.g., human immunodeficiency virus [HIV]), the order of the stages differs from that in this cartoon (see Fig. 13.2). Some viruses lack stages shown here (e.g., release), whereas other viruses have additional stages. (Modified from Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Inhibition of Viral Attachment and Entry
Inhibition of attachment and entry prevents all subsequent steps in virus infection and permits the virion to be cleared by immune and other mechanisms. There have been two general approaches for drugs that inhibit attachment and entry, which thus far have resulted in two approved anti-HIV drugs: maraviroc and enfuvirtide. Both of these drugs have unusual properties; maraviroc targets a host protein, whereas enfuvirtide is a peptide.
CCR5 Blockers
Maraviroc (Fig. 13.3) is a CCR5 blocker; it inhibits binding of HIV to the CCR5 co-receptor. CCR5 is the co-receptor used by the most commonly transmitted HIV-1 strains (R5 tropic), which predominate during the early stages of HIV-1 infection.26 There are several attractive features to CCR5 as a target for anti-HIV drugs: Knockout mice lacking CCR5, and humans with homozygous deletions in the gene encoding CCR5 (CCR5Δ32) lack detectable abnormalities, suggesting that blocking this host function might not have adverse consequences.38 Certain of these CCR5Δ32 homozygotes who have been highly exposed to HIV have remained uninfected, and their cells are resistant to R5-tropic strains of HIV in vitro.221 Blockade of CCR5 with a natural ligand (e.g., RANTES) or antibody inhibits R5-tropic HIV replication (e.g.,56). These observations suggested that
blocking CCR5 could be efficacious against R5-tropic HIV without causing much toxicity.
blocking CCR5 could be efficacious against R5-tropic HIV without causing much toxicity.
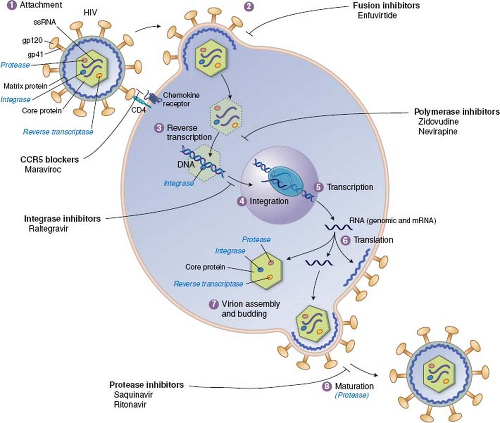 Figure 13.2. Stages of HIV infection blocked by different classes of antiviral drugs. The lifecycle of HIV is cartooned, showing the following. 1: Virus attachment is dependent on binding interactions between viral gp160 (composed of gp41 and gp120 proteins) and host cell CD4 and certain chemokine receptors. 2: Fusion of the viral membrane (envelope) with the host cell plasma membrane allows the human immunodeficiency (HIV) genome complexed with certain virion proteins to enter the host cell. 3: Uncoating permits the single-stranded RNA (ssRNA) HIV genome to be copied by reverse transcriptase into double-stranded DNA. 4: The HIV DNA is integrated into the host cell genome, in a reaction that depends on HIV-encoded integrase. 5: Gene transcription and posttranscriptional processing by host cell enzymes produce genomic HIV RNA and viral messenger RNA (mRNA). 6: The viral mRNA is translated into proteins on host cell ribosomes. 7: The proteins and genomic RNA assemble into immature virions that bud from the host cell membrane. 8: The virions undergo proteolytic cleavage, maturing into fully infective virions. The steps at which classes of anti-HIV drugs act (attachment, fusion, reverse transcription, integration, and maturation) are indicated, with examples from specific drug classes provided. (Modified from Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Maraviroc was discovered using high-throughput screening for small molecules that block binding of CCR5 to a natural ligand, followed by medicinal chemistry efforts to improve upon a hit from the screen.83 Modeling and mutational studies suggest that maraviroc and several other CCR5 blockers bind to a pocket on CCR5 formed by transmembrane domains,195 and allosterically inhibit binding to HIV gp120, thereby blocking HIV entry (Fig. 13.3).9,83,112,374 R5 tropic HIV mutants resistant to CCR5 blockers including maraviroc contain altered gp120 molecules that are able to bind to the drug-bound form of CCR5.366,374 Maraviroc was approved in 2007 for use in combination with other anti-HIV drugs in patients with R5-tropic
strains. Therefore, patients must be tested to determine if they harbor virus that utilizes a different co-receptor (i.e., CXCR4). Maraviroc treatment failures are more frequently attributable to preexisting minority populations of CXCR4-tropic virus than to selection of R5-tropic virus that is resistant to maraviroc.259 Resistance to CCR5 blockers has not resulted thus far from R5-tropic virus evolving to use a different co-receptor. Nevertheless, this possibility and the effects of antagonizing a host function remain potential issues for the long-term clinical use of maraviroc and other CCR5 blockers, which are in development (e-Table 13.1).
strains. Therefore, patients must be tested to determine if they harbor virus that utilizes a different co-receptor (i.e., CXCR4). Maraviroc treatment failures are more frequently attributable to preexisting minority populations of CXCR4-tropic virus than to selection of R5-tropic virus that is resistant to maraviroc.259 Resistance to CCR5 blockers has not resulted thus far from R5-tropic virus evolving to use a different co-receptor. Nevertheless, this possibility and the effects of antagonizing a host function remain potential issues for the long-term clinical use of maraviroc and other CCR5 blockers, which are in development (e-Table 13.1).
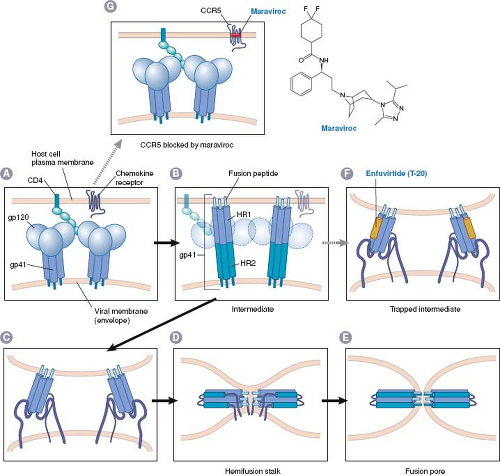 Figure 13.3. Model for HIV gp41-mediated fusion and maraviroc and enfuvirtide (T-20) action. A: Human immunodeficiency virus (HIV) glycoproteins exist in trimeric form in the viral envelope. Each gp120 molecule is depicted as a ball attached noncovalently to gp41. The CD4 receptor and a chemokine receptor that serves as a co-receptor in the host cell plasma membrane are shown. B: The binding of gp120 to CD4 and the co-receptor causes a conformational change in gp41 that exposes the fusion peptide, heptad-repeat region 1 (HR1) and heptad-repeat region 2 (HR2). The fusion peptide inserts into the host cell plasma membrane. C: gp41 undergoes further conformational changes, characterized by unfolding and refolding of the HR2 segments. D: Completed refolding of the HR regions creates a hemifusion stalk in which the outer leaflets of the viral and host cell membranes are fused. E: Formation of a complete fusion pore allows viral entry into the host cell. F: Enfuvirtide (T-20) is a synthetic peptide that mimics HR2 and binds to HR1. It prevents the HR1–HR2 interaction (dashed arrow). The drug, therefore, traps the virus–host cell interaction at the attachment stage, preventing membrane fusion and viral entry. G: Maraviroc is a small molecule antagonist of the CCR5 chemokine receptor; the drug blocks cellular infection of HIV strains that use CCR5 for attachment and entry (dashed arrow from A to G. The structure of maraviroc is shown). (From Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Fusion Inhibitors: Enfuvirtide
Although compounds that bind to virus particles and block attachment and entry have been investigated in the laboratory and clinic for decades (e.g., the anti-picornavirus drug, pleconaril), enfuvirtide was the first drug approved for clinical use that acts this way. This agent was discovered by a rational, directed approach that examined the ability of peptides to inhibit HIV infection in cell culture.385,386 The peptide that was most potent (T-20) is similar to a segment of gp41, the HIV protein that mediates membrane fusion. The proposed mechanism for gp41-mediated membrane fusion and T-20 action is illustrated in Figure 13.3: The gp41 protein in the virion is ordinarily trapped in a metastable conformation so that it cannot fuse membranes or bind T-20. Binding of HIV to its cellular receptors triggers a conformational change that exposes the segment of gp41 that is thought to insert into host cell membranes (fusion peptide), a heptad repeat region (HR-1), and the segment that T-20 resembles, which is a second heptad repeat (HR-2). The gp41 then rearranges so that HR-1 and HR-2 bind to each other. If the fusion peptide has inserted into the host cell membrane, this refolding brings the virion envelope and the cell membrane in proximity, thereby allowing membrane fusion to occur (by mechanisms that remain incompletely understood). When T-20 is present, however, it binds to HR-1 and prevents the refolding process, thereby preventing fusion of the HIV envelope with the host cell membrane.
Although this model requires an exogenous peptide to compete with an intramolecular interaction to bind to a target that is available only transiently, it is supported by a number of observations. These include the crystal structure of the rearranged form of gp41, which shows the HR-1–HR-2 interaction41,357,373 and evidence that T-20 interacts with HR-1 containing peptides.42,205,384 Genetic studies show that HR-1 and HR-2 are crucial for fusion.88,383 Perhaps the strongest evidence that T-20 inhibits HIV replication by binding to HR-1 is that HIV mutations that confer resistance to T-20 alter HR-1 residues, and that these alterations weaken binding of T-20 to HR-1.312
Not only is the mechanism of T-20 remarkable, but so is its development into a drug. Because it is a peptide, there were major obstacles to its large-scale synthesis and manufacture, and outpatients must mix the drug and inject it themselves. Regardless, it proved that inhibitors of attachment and entry can be effective antiviral agents.
Inhibition of Viral Uncoating: Adamantane Derivatives
The adamantanes, amantadine and rimantadine (Fig. 13.4), are active exclusively against influenza A virus (and not influenza B or C viruses). In most influenza A virus strains, these drugs inhibit virus uncoating. It took decades to identify the target of these drugs. A key finding142,143 was that amantadine has an unusual dose–response curve, at least for some strains of influenza A virus. It inhibits viral replication at concentrations of approximately 1 μM, which are those attained in patients treated with the drugs; however, at somewhat higher concentrations, it loses antiviral activity. It then regains antiviral activity at concentrations greater than 100 μM. Hay and colleagues142 then showed that mutants that could replicate in 5 μM amantadine were resistant due to point mutations within the M segment of influenza RNA gene, in an open reading frame that had only recently been recognized. This open reading frame encodes a small membrane protein known as M2 (the M segment also encodes the long-recognized matrix [M1] protein.) Subsequent biochemical and electrophysiologic experiments showed that the M2 protein forms a tetrameric, pH-gated channel for H+ ions and that amantadine and rimantadine block this channel.17,89,163,289,349
The manner in which amantadine and rimantadine block the M2 ion channel has been investigated intensively. Viral mutations conferring resistance to these drugs alter any of five residues in the transmembrane segment of the protein that are predicted to be within the pore of the channel, suggesting that the drugs bind to these residues and “plug” the channel.141 Structural studies of drug-bound versions of M2 followed by electrophysiologic assays using Xenopus oocytes and assays of engineered viral mutants support this suggestion.36,180,270,343 However, evidence from nuclear magnetic resonance (NMR) studies and liposomal proton flux assays supported the suggestion that these drugs do not act as “plugs,” but instead bind to the outside of the channel and stabilize its closed conformation.287,330 This controversy appears to have been resolved in favor of the pore-binding site being the one that mediates drug action, with the NMR structure of rimantadine bound within the pore of a drug-sensitive chimeric M2 derived from influenza A and B viruses.286
How would blocking an ion channel lead to inhibition of uncoating? A reasonable but not completely verified model is diagrammed in Figure 13.4. Influenza A virus attaches to sialic acid moieties on cell surface glycoproteins, and then, bound to its receptor, is internalized, surrounded by a cell membrane–bounded compartment, the endosome, which normally participates in membrane protein recycling. As part of its normal function, the endosome becomes acidified. During influenza virus entry, this reduction in pH causes a conformational change in the virion hemagglutinin protein, and fusion of the virion envelope and the endosomal membrane via a mechanism similar to that of HIV gp41 (Fig. 13.3). By itself, this action would release viral ribonucleoprotein into the cytoplasm. However, in the presence of amantadine or rimantadine, the matrix protein, M1, does not dissociate from the ribonucleoprotein, which remains in the cytoplasm instead of entering the nucleus.34,236 Low pH can promote the dissociation of M1, and allow nuclear entry of the ribonucleoprotein.33 Therefore, M2 in the virion envelope functions to let H+ ions from the acidified endosome enter the virion, and dissociate M1 from the ribonucleoprotein. Amantadine and rimantadine would block the entry of H+ ions, thereby preventing this uncoating event.
With certain influenza A virus strains, amantadine and rimantadine also block late events in virus replication, in particular,
maintenance of hemagglutinin in a proper conformation for infectivity.141
maintenance of hemagglutinin in a proper conformation for infectivity.141
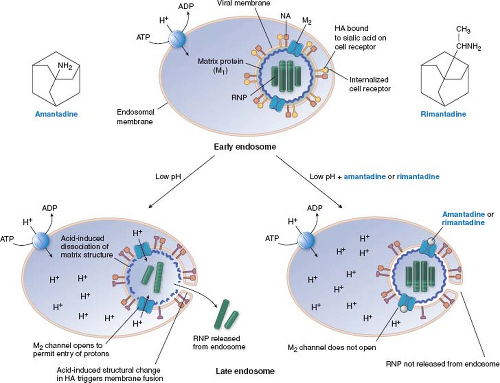 Figure 13.4. Model for uncoating of influenza A virus and effect of amantadine or rimantadine. The structures of amantadine and rimantadine are shown. The process of influenza A virus uncoating and drug action is depicted. The virus enters host cells by receptor-mediated endocytosis (not shown) and is contained within an early endosome. The early endosome contains a H+-ATPase that acidifies the endosome by pumping protons from the cytosol into the endosome. A low pH-dependent conformational change in the envelope protein, hemagglutinin (HA), triggers fusion of the viral membrane with the endosomal membrane. This fusion event, however, is not sufficient to cause viral uncoating. In addition, protons from the late endosome must enter the virion through the M2 proton channel, which is a virion envelope protein that opens in response to acidification (pH-gated ion channel). The entry of protons into the virion causes dissociation of the M1 matrix protein from the influenza A ribonucleoprotein (RNP), releasing it into the cytoplasm. The dissociation of matrix is required for RNP to be transported to the nucleus, where it is transcribed and replicated. Amantadine and rimantadine block M2 ion channel function and thereby inhibit acidification of the interior of the virion, matrix dissociation, and uncoating. NA, neuraminidase. (Modified from Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Influenza virus mutants that are resistant to amantadine and rimantadine due to altered M2 have been assayed in animal models including mice and ferrets, and show little or no diminution in their replication or pathogenesis.15,273,355 This correlates well with their behavior in human infections.144 Since the discoveries of the mechanism of adamantanes and the function of M2, a number of other viruses have been found to encode proteins that can form ion channels. These also might serve as good drug targets.
Inhibition of Viral Gene Expression: NS3/4A Protease Inhibitors
Viral gene expression entails not only the transcription of the viral genome into mRNA, the translation of mRNA into protein, and the stabilities of RNA and protein gene products, but can also include a variety of processing events including capping, splicing, and polyadenylation of viral mRNAs, and proteolytic cleavage of viral polyproteins into their individual protein units. Many viruses encode functions that execute or abet one or more of these steps in gene expression, and much
effort has gone into targeting these functions for development of antiviral drugs. In particular, viral proteases that generate individual viral proteins have been targeted. The activity of one of these, the HIV protease, is not required until the viral particle is fully assembled, and thus will be discussed under the heading Inhibition of Viral Assembly and Maturation below. The activity of the HCV NS3/4A protease, however, is required for proper expression of viral gene products for essentially all subsequent steps in viral infection. In 2011, the first drugs that target this stage of viral replication were approved by the U.S. Food and Drug Administration (FDA). These two drugs—telaprevir and boceprevir (Fig. 13.5)—are peptidomimetics that inhibit the NS3/4A protease.
effort has gone into targeting these functions for development of antiviral drugs. In particular, viral proteases that generate individual viral proteins have been targeted. The activity of one of these, the HIV protease, is not required until the viral particle is fully assembled, and thus will be discussed under the heading Inhibition of Viral Assembly and Maturation below. The activity of the HCV NS3/4A protease, however, is required for proper expression of viral gene products for essentially all subsequent steps in viral infection. In 2011, the first drugs that target this stage of viral replication were approved by the U.S. Food and Drug Administration (FDA). These two drugs—telaprevir and boceprevir (Fig. 13.5)—are peptidomimetics that inhibit the NS3/4A protease.
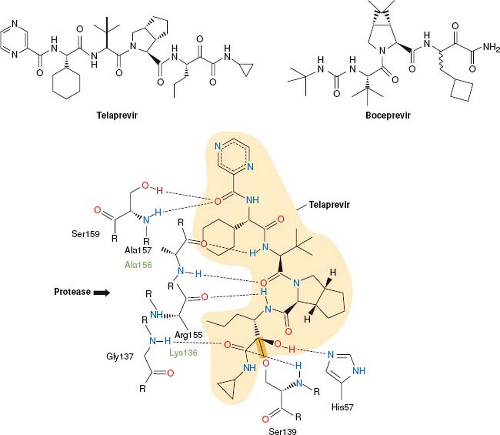 Figure 13.5. Anti-HCV (hepatitis C virus) protease inhibitors. The structures of telaprevir (left) and boceprevir (right) are shown at the top of the figure. A schematic of the interaction of telaprevir with the HCV NS3/4A protease is shown at the bottom of the figure. Important hydrogen-bonding interactions between protease residues and drug are shown with dashed lines, whereas protease residues that make important hydrophobic interactions are indicated in green. The covalent bond formed between telaprevir and serine 139 of NS3 is highlighted in yellow. (Courtesy of Ayesegui Özen and Celia Schiffer, based on the crystal structure of telaprevir bound to NS3/4A. From Romano KP, Ali A, Soumana D, et al. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog 2012;8:e1002832.) |
Following entry and uncoating of HCV, its genome serves as an mRNA that is translated into a very large (∼3,000 residue) polyprotein. This polyprotein is processed into 10 individual proteins by cellular and viral proteases. One viral protease, which is especially important for generating nonstructural (NS) proteins, is NS3. NS3 contains an N-terminal protease domain and a C-terminal helicase domain. A second protein, NS4A, forms a complex with NS3 (NS3/NS4A), and enhances its protease activity. During polyprotein processing, NS3 autoproteolytically cleaves between NS3 and NS4A, and then cleaves to generate NS4A (its co-factor), NS4B, NS5A, and NS5B. The NS proteins are essential for HCV genome replication and production of infectious virus. These proteins, especially NS3, are also thought to counteract the innate immune response.109
Given the success of drugs that inhibit the HIV protease (see below), investigators launched efforts to develop drugs that inhibit the NS3 protease. As described below in more detail for the HIV protease inhibitors, the development
process was iterative involving the synthesis of candidate inhibitors, solving the structures of these inhibitors bound to the protease, and then designing new compounds with higher potency and better pharmacological properties based on those structures.213,266 Several features of the HCV NS3/4A protease and its inhibitors differ from those of their HIV counterparts, however. The HIV protease is an aspartyl protease, whereas the NS3/4A protease is a serine protease. The HIV protease’s substrate binding pocket is deep, so that there are multiple opportunities for interactions with a small molecule inhibitor, whereas the NS3/4A binding pocket is shallow, with relatively few obvious sites for binding a small molecule. Despite this obstacle, structure-based medicinal chemistry efforts identified sites on NS3 that could mediate tight and selective binding of inhibitors (Fig. 13.5).213,266 The anti-HIV protease inhibitors are largely symmetrical, mimicking the protein substrate on both sides of the protease cleavage site, whereas the anti-NS3/4A inhibitors are relatively asymmetric, mainly mimicking the N-terminal product of protease cleavage. The anti-HIV protease inhibitors contain CHOH moieties that mimic the transition state of protease catalysis to achieve tight, but noncovalent binding to the enzyme (transition state analogs). In contrast, the anti-NS3/4A inhibitors contain ketoamide groups that react with the catalytic serine of the enzyme to form a covalent bond with the enzyme (Fig. 13.5), which only very slowly reverses, resulting in time-dependent inhibition of the enzyme (mechanism-based inhibitors) and inhibition constants of ∼10 nM.213,266
process was iterative involving the synthesis of candidate inhibitors, solving the structures of these inhibitors bound to the protease, and then designing new compounds with higher potency and better pharmacological properties based on those structures.213,266 Several features of the HCV NS3/4A protease and its inhibitors differ from those of their HIV counterparts, however. The HIV protease is an aspartyl protease, whereas the NS3/4A protease is a serine protease. The HIV protease’s substrate binding pocket is deep, so that there are multiple opportunities for interactions with a small molecule inhibitor, whereas the NS3/4A binding pocket is shallow, with relatively few obvious sites for binding a small molecule. Despite this obstacle, structure-based medicinal chemistry efforts identified sites on NS3 that could mediate tight and selective binding of inhibitors (Fig. 13.5).213,266 The anti-HIV protease inhibitors are largely symmetrical, mimicking the protein substrate on both sides of the protease cleavage site, whereas the anti-NS3/4A inhibitors are relatively asymmetric, mainly mimicking the N-terminal product of protease cleavage. The anti-HIV protease inhibitors contain CHOH moieties that mimic the transition state of protease catalysis to achieve tight, but noncovalent binding to the enzyme (transition state analogs). In contrast, the anti-NS3/4A inhibitors contain ketoamide groups that react with the catalytic serine of the enzyme to form a covalent bond with the enzyme (Fig. 13.5), which only very slowly reverses, resulting in time-dependent inhibition of the enzyme (mechanism-based inhibitors) and inhibition constants of ∼10 nM.213,266
Both boceprevir and telaprevir exhibit submicromolar potencies (concentrations that inhibit replication by 50%) for inhibition of replication of HCV genomes in a cell-based replicon system.214,229 Interestingly, telaprevir was considerably less potent in these assays than a macrocyclic NS3/4A inhibitor, BILN 2061, which binds tightly but noncovalently; yet both drugs eliminated HCV replicons (104-fold reductions) from cells at similar concentrations, which may relate to the very slow reversibility of telaprevir.214 This observation helps to illustrate the concept that efficacy (how much viral replication is reduced) is likely to be more important than potency in antiviral development. Both boceprevir and telaprevir inhibit HCV genome replication, at least additively in combination with interferon α (IFN-α).214,229 These findings are important due to the use of boceprevir and telaprevir in combination with IFN-α for HCV therapy, and they may relate to a role for NS3 in counteracting IFN action.109 Once a suitable cell culture system that supports complete replication of HCV was devised, it was shown that NS3/4A inhibitors block production of infectious virus.215 Finally, as was first strikingly demonstrated with BILN 2061, NS3/4A inhibitors can drastically reduce levels of HCV in the plasma of patients.197
HCV replication is rapid and error-prone; therefore, it is not surprising that resistance to NS3/4A protease inhibitors arises rapidly both in cell culture systems and in patients.82 Indeed, resistant variants preexist in untreated patients.14 The different subtypes of HCV complicate HCV drug resistance, with protease inhibitors having different potencies against different subtypes, and with different subtypes requiring different numbers of mutations to achieve resistance.132
Several mutations confer resistance to boceprevir and telaprevir, with some altering NS3 residues that make direct contacts with drug and others altering more distant residues that contact amino acids close to the drug, such as the catalytic serine.132 Certain mutations confer differing degrees of resistance to the two drugs. Notably, some mutations that confer resistance to boceprevir and telaprevir do not confer resistance to newer macrocyclic NS3/4A inhibitors that are in development (e-Table 13.1), and certain mutations that are resistant to the macrocyclic inhibitors do not confer resistance to boceprevir and telaprevir. However, other mutations confer resistance to both classes of NS3/4A drugs.132 The effects of the different mutations on fitness vary as measured in cell culture systems for genome replication and production of infectious virus or in human hepatocyte chimeric mice, with some mutations being clearly impaired and others having very little impact.158,334,363 Mathematical modeling of HCV infections in humans suggests that at least some protease inhibitor–resistant mutations retain substantial fitness.316 These features of resistance to boceprevir and telaprevir are an impetus for continued development of anti-HCV drugs that can be used in combination (e-Table 13.1).
Inhibition of Viral Genome Replication
Most antiviral drugs inhibit viral genome replication, and nearly all of these inhibit a viral polymerase. Those viruses whose genome replication has been successfully targeted to yield FDA-approved drugs include certain human herpesviruses, the retrovirus HIV, and the hepadnavirus HBV. Several inhibitors of the polymerase of the flavivirus HCV are in clinical development. Most of the approved drugs are nucleoside analogs (Figure 13.6). Some of these nucleoside analogs actually mimic nucleoside monophosphates, so they are actually nucleotide analogs. A few approved drugs are nonnucleoside inhibitors of DNA polymerase or reverse transcriptase that act by binding at a site other than the dNTP-binding site, and one approved drug inhibits HIV integrase.
Nucleoside Analogs
Nucleoside analogs that clearly act by inhibiting viral polymerases are approved for treatment of herpesviruses, HBV, and HIV, and several are in development for treatment of HCV. Another nucleoside analog, ribavirin, is used clinically against HCV and respiratory syncytial virus, but its mechanism of action against those viruses is not well understood (see below). All nucleoside and nucleotide analogs must be activated by phosphorylation, usually to the triphosphate form, to exert their effect. Phosphorylated nucleoside analogs inhibit polymerases by competing with the natural substrate (dNTP for DNA polymerase) and are usually also incorporated into the growing nucleic acid chain, where they often terminate elongation. Either or both of these features—inhibition and incorporation—can be important for antiviral activity.
The more efficiently cellular enzymes phosphorylate the nucleoside analog and the more potently the phosphorylated forms act against cellular enzymes, the more toxic the nucleoside analog will be. Selectivity, therefore, is dependent upon how much more potently and effectively viral genome replication is inhibited than are cellular functions, and for the herpesviruses, how much more efficiently viral enzymes phosphorylate the drug than do cellular enzymes.
Anti-Herpesvirus DNA Polymerase Inhibitors
The various human herpesviruses encode both kinases and DNA polymerases. The kinases are usually not essential for viral
replication, but the catalytic subunits of the polymerases (Pols) are. These enzymes differ enough from their cellular counterparts to permit development of selective antiviral nucleoside analog.
replication, but the catalytic subunits of the polymerases (Pols) are. These enzymes differ enough from their cellular counterparts to permit development of selective antiviral nucleoside analog.
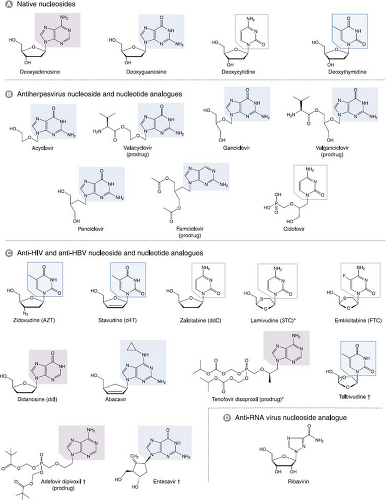 Figure 13.6. Structures of antiviral nucleoside analogues. A: The deoxynucleosides used as precursors for DNA synthesis are depicted in their anti configurations. The base moieties are shaded. B: Anti-herpesvirus nucleoside analogs mimic the deoxynucleoside, deoxyguanosine, except for cidofovir, which mimics the deoxynucleotide dCMP. The compounds shown here all contain acyclic moieties that mimic deoxyribose. Valacyclovir and famciclovir are prodrugs of acyclovir (ACV) and penciclovir, respectively. C: Anti-HIV (human immunodeficiency virus) and anti-HBV (hepatitis B virus) nucleoside analogs mimic dT, dC, dA, and dG except for tenofovir, and adefovir, which mimic dAMP. All of the analogs contain a base found in A, except for didanosine, which mimics deoxyinosine and is converted to dideoxyadenosine, and abacavir, which contains a cyclopropyl-modified guanine. Tenofovir and adefovir are shown as their disoproxil and dipivoxil prodrugs, respectively. Lamivudine, emtricitabine, and telbivudine are L-stereoisomers, whereas the other nucleoside analogs and dT, dC, dA, and dG are D-stereoisomers. All of the compounds shown except entecavir contain deoxyribose mimics that lack the equivalent of a 3′-hydroxyl, and thus are obligate chain terminators. The anti-HIV nucleoside analogs, lamivudine and tenofovir(*), and telbivudine, adefovir, and entecavir (†) are approved for use against HBV. D: Ribavirin, which contains a purine mimic attached to ribose, is approved for use against the RNA viruses hepatitis C virus and respiratory syncytial virus. (Modified from Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
A number of antiviral nucleoside analogs including vidarabine, idoxuridine, and trifluridine, were developed and used against HSV infections. However, these drugs have been superseded by more selective compounds, and their mechanisms will not be discussed further.
Acyclovir and Related Drugs
ACV is the paradigmatic antiviral nucleoside analog that illustrates many of the principles of nucleoside analogs. It consists of a guanine base attached to an acyclic sugar-like molecule (Fig. 13.6). ACV and its more orally available valine ester, valacyclovir, and the related drugs, penciclovir and its orally available derivative, famciclovir (structures in Fig. 13.6) have similar mechanisms of action against HSV and VZV. ACV was originally synthesized at Burroughs Wellcome as part of a program to discover adenosine deaminase inhibitors, and only later was it tested for antiviral activity. The mechanism of ACV action91 is presented in Figure 13.7. The TK encoded by HSV and VZV phosphorylates ACV, despite its differences from the natural substrate of this enzyme, thymidine (also known as deoxythymidine; see Fig. 13.6). Crystal structures of HSV TK in complex with thymidine or ACV29,40,387 show that the guanine moiety of ACV interacts with the same residues as does the thymine moiety of thymidine, albeit with differences in water-mediated binding and bond angles. Despite this, ACV is not a particularly good substrate for HSV or VZV TK. Nevertheless, no mammalian enzyme phosphorylates ACV nearly as efficiently as the HSV and VZV TKs. Accordingly, HSV- and VZV-infected cells contain 30- to 100-fold more phosphorylated ACV than do uninfected cells, which accounts for much of ACV’s antiviral selectivity.
Phosphorylation of ACV by the viral TK produces the compound ACV-monophosphate, which is converted sequentially to ACV-diphosphate and ACV-triphosphate (ACV-TP) by cellular enzymes.252,253 ACV-TP then inhibits HSV and VZV DNA polymerases. Inhibition of HSV DNA polymerase in vitro is a three-step process (Fig. 13.7).300 In the first step, ACV-TP competitively inhibits deoxyguanosine triphosphate (dGTP) incorporation (high concentrations of dGTP can reverse inhibition at this early step). Next, ACV-TP acts as a substrate and is incorporated into the growing DNA chain opposite a C residue. Finally, the polymerase translocates to the next position on the template, but cannot add a new deoxynucleoside triphosphate because ACV contains no 3′-hydroxyl; hence ACV-TP is an obligate chain terminator. Provided that the next dNTP is present, the viral polymerase freezes at this final step in a “dead-end complex,” leading to apparent inactivation of the enzyme. There is biochemical evidence for selectivity at each of these steps, especially the third.174,235,299 The results of enzymological studies of viral polymerases from ACV-resistant mutants167 argue that in infected cells, as in vitro, ACV-TP is not simply a competitive inhibitor of viral DNA polymerase, but rather that its incorporation is crucial, consistent with the three step model (Fig. 13.7).
Stocks of most HSV-1 strains contain ACV-resistant mutants at a level of about 0.01% to 0.1% (reviewed in 57). For at least certain HSV-2 strains, mutation frequencies are an order of magnitude higher.323 Compared with the drugs discussed so far, there is considerable variety in ACV-resistant mutants. ACV selects mainly for tk mutants60,324,354 that ablate (TK-negative) or reduce (TK-partial) TK activity or alter TK so that it fails to phosphorylate ACV, but continues to phosphorylate dT (TK-altered). ACV-resistance mutations can also alter the viral polymerase to be less inhibited by the drug (reviewed in 199). Crystallographic studies have provided details on how tk and pol mutations affect binding and catalysis.29,40,102,167,222,387
Nearly all ACV-resistant HSV mutants exhibit some degree of attenuation in assays of pathogenesis in animal models (reviewed in 58 and 199). TK-negative mutants are generally the most attenuated, especially for replication in sensory ganglia and reactivation from latency in those ganglia. However, certain clinical isolates are able to reactivate despite being TK negative.123,164 TK-partial mutants are less attenuated. As little as 5% to 10% of wild-type levels of TK activity permit normal ganglionic replication and reactivation from latency,44,59 and very low levels of TK produced via these translational mechanisms can suffice to permit some reactivation from latency.19,123 Interestingly, certain nonsense and frameshift mutations that might be expected to inactivate TK do not do so. This “leakiness” can result from translational mechanisms, including reinitiation and ribosomal frameshifting, that permit low levels of TK despite these mutations.124,165,171,176 Therefore, several mutants that would be expected to be TK negative are actually TK partial. TK-altered and pol mutants include the most pathogenic drug-resistant mutants, but some pol mutants are highly attenuated.
This picture becomes even more complicated when mixtures of drug-resistant mutants with each other or with drug-sensitive virus are considered, as these can complement each other for both drug resistance and pathogenicity.58,199 In addition, tk frameshift mutations in homopolymeric sequences, which are the most common drug-resistance mutations, tend to revert, sometimes at remarkably high rates, resulting in mixed populations that reactivate from latency.122,124,326 Therefore, it is important to consider heterogeneity as a factor influencing drug resistance and pathogenicity.
Ganciclovir and Valganciclovir
Although ACV has been highly successful in treating HSV and VZV disease, it is much less potent against HCMV,74 which causes serious diseases in immunocompromised persons. This is primarily because much less phosphorylated ACV accumulates in HCMV-infected cells than in HSV- or VZV-infected cells.106 This helped spur the development of ganciclovir. Ganciclovir is much more potent against HCMV than is ACV47,336 and was the first drug approved for use against HCMV. Ganciclovir is now also produced as the more orally available agent valganciclovir (see Fig. 13.6).
Ganciclovir, unlike ACV, but like penciclovir, has the equivalent of a 3′-OH moiety (see Fig. 13.6). Much as phosphorylated ACV accumulates in HSV-infected cells, phosphorylated ganciclovir accumulates in HCMV-infected cells.21,104 However, as HCMV does not encode a TK, this phosphorylation was initially thought to be due to induction of a host cell kinase. However, studies of a ganciclovir-resistant mutant led to the discovery that an unusual HCMV protein kinase (UL97) phosphorylates ganciclovir in infected cells.152,219,248,350,356 Cellular kinases can convert ganciclovir monophosphate to ganciclovir triphosphate. Like ACV triphosphate, ganciclovir
triphosphate is both a selective competitive inhibitor and substrate for viral DNA polymerase.21,115,233,235,303,339 Unlike ACV triphosphate, ganciclovir triphosphate is not an obligate chain terminator. Nevertheless, after incorporating ganciclovir monophosphate, HCMV DNA polymerase can stall after incorporating one additional nucleotide.303 However, biochemical and genetic studies indicate that selectivity at the phosphorylation step and the DNA polymerase inhibition step for HCMV are not as great as the selectivity of ACV against HSV. Accordingly, the drug is more toxic than ACV. Toxicity is most commonly manifested in patients as bone marrow suppression, especially neutropenia.267
triphosphate is both a selective competitive inhibitor and substrate for viral DNA polymerase.21,115,233,235,303,339 Unlike ACV triphosphate, ganciclovir triphosphate is not an obligate chain terminator. Nevertheless, after incorporating ganciclovir monophosphate, HCMV DNA polymerase can stall after incorporating one additional nucleotide.303 However, biochemical and genetic studies indicate that selectivity at the phosphorylation step and the DNA polymerase inhibition step for HCMV are not as great as the selectivity of ACV against HSV. Accordingly, the drug is more toxic than ACV. Toxicity is most commonly manifested in patients as bone marrow suppression, especially neutropenia.267
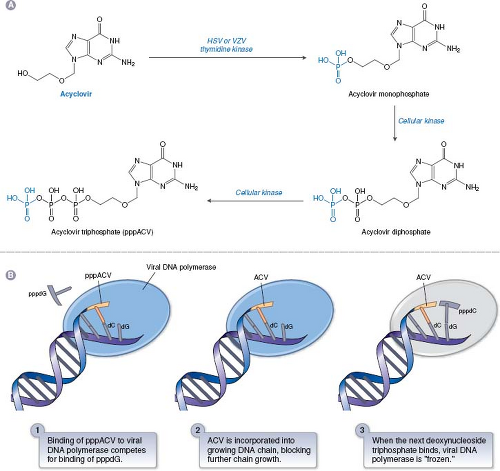 Figure 13.7. Mechanism of action of acyclovir (ACV). A: ACV is selectively phosphorylated by herpes simplex virus (HSV) or varicella zoster virus (VZV) thymidine kinase to generate ACV monophosphate. Host cellular enzymes then sequentially phosphorylate the drug monophosphate to the diphosphate and triphosphate (pppACV) forms. B: ACV triphosphate has a three-step mechanism of inhibition of herpesvirus DNA polymerase in vitro: (1) The drug triphosphate acts as a competitive inhibitor of deoxyguanosine triphosphate (dGTP) (pppdG) binding; (2) the drug triphosphate acts as a substrate and is incorporated into the growing DNA chain across from dC in the template, terminating elongation; and (3) the polymerase becomes trapped on the ACV-terminated DNA chain when the deoxynucleotide triphosphate (dNTP) binds (here shown as pppdC, dCTP, which would be templated by dG). (From Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Most ganciclovir-resistant mutants contain UL97 mutations, but these have a limited distribution in the gene.11,118
Unlike HSV TK, HCMV UL97 protein kinase is very important for viral replication (i.e., null mutants are much less fit296). It seems likely that clinical UL97 drug-resistance mutations would affect recognition of ganciclovir without gravely compromising activity on the physiologic protein substrates of UL97 such as retinoblastoma protein and lamin A.135,169,185 However, this has not yet been demonstrated. Numerous different pol mutations have been shown to confer ganciclovir resistance (reviewed in 11 and 118). Interestingly, some lie in or near regions not altered in ACV-resistant HSV mutants, but in regions that are thought to be important for 3′ to 5′ exonuclease activity. How these mutations confer ganciclovir resistance is not yet known.
Unlike HSV TK, HCMV UL97 protein kinase is very important for viral replication (i.e., null mutants are much less fit296). It seems likely that clinical UL97 drug-resistance mutations would affect recognition of ganciclovir without gravely compromising activity on the physiologic protein substrates of UL97 such as retinoblastoma protein and lamin A.135,169,185 However, this has not yet been demonstrated. Numerous different pol mutations have been shown to confer ganciclovir resistance (reviewed in 11 and 118). Interestingly, some lie in or near regions not altered in ACV-resistant HSV mutants, but in regions that are thought to be important for 3′ to 5′ exonuclease activity. How these mutations confer ganciclovir resistance is not yet known.
Cidofovir and Other Nucleotide Analogs
Cidofovir, a phosphonate-containing acyclic cytosine analog, is approved for use against HCMV, and represents a variation on the mechanism of action of nucleoside analogs. Cidofovir, with its phosphonate group, is a nucleotide analog that mimics deoxycytidine monophosphate (dCMP) (see Fig. 13.6). This charged moiety likely accounts for the relatively poor uptake of cidofovir into cells,161 and poor oral bioavailability. Once inside the cell, cidofovir is metabolized via cellular enzymes to its monophosphate (akin to a diphosphate) and diphosphate (akin to a triphosphate), and to a third phosphorylated form that contains a choline adduct.52,161 These phosphorylated forms have very long intracellular half-lives, which may be due in part to the choline metabolite serving as a reservoir.1,161 This property contributes to the prolonged antiviral activity of cidofovir,260 which provides the therapeutic advantage of infrequent dosing. In addition, because cidofovir uses cellular kinases for its phosphorylation, it is active against UL97 mutants that are resistant to ganciclovir,225 although this also removes the selectivity gained by requiring a viral kinase for efficient phosphorylation.
The diphosphorylated form of cidofovir is an analog of dCTP that inhibits HCMV DNA polymerase more potently than it does various cellular DNA polymerases.48,161,394 It is also incorporated into DNA, which slows elongation, but does not result in chain termination unless two cidofovir residues are incorporated in a row.393,394 Many of the pol mutations that confer resistance to ganciclovir confer resistance to cidofovir.
Two related phosphonate-containing drugs are the acyclic deoxyadenosine monophosphate analogues, tenofovir, which is used against HIV and HBV, and adefovir, which has been used against HBV (see Fig. 13.6). The mechanisms of action of these drugs against their respective viruses are similar to those of cidofovir against HCMV, except that these drugs are chain-terminators of DNA synthesis. (See below for discussions of nucleoside analogs active against HIV and HBV.) These two drugs are administered as orally available prodrugs (Fig. 13.6). In contrast, there is currently no such approved prodrug for cidofovir, which must be administered intravenously. Orally available prodrugs of cidovir are being developed with the added potential benefit of activity against poxviruses, adenoviruses, polyomaviruses, and papillomaviruses166 (e-Table 13.1).
Nonnucleoside Inhibitors of Herpesvirus DNA Polymerase: Foscarnet
As described above, nucleoside analogs can inhibit cellular as well as viral enzymes, and viruses can mutate to resist these drugs. As a result, efforts have been made to discover compounds that might inhibit viral polymerases by other mechanisms. The first of these to be approved for clinical use was foscarnet (phosphonoformic acid, PFA; structure in Fig. 13.8). Although it is not orally available so that intravenous administration is required, and it is nephrotoxic, it is approved for treatment of severe HSV, VZV, and HCMV infections that are resistant to front-line drugs.
Foscarnet is an analog of pyrophosphate, which is a product of polymerization of nucleic acids. Unlike the nucleoside analogs described above, it does not require activation by either cell or viral enzymes but rather inhibits HCMV DNA polymerase directly and selectively. Inhibition is not competitive with deoxynucleoside triphosphates. Rather, it appears that foscarnet acts as a product analog, preventing normal pyrophosphate release so the polymerase cannot complete the catalytic cycle.93,271 The structure of foscarnet bound to a DNA polymerase that is a chimera of a bacteriophage DNA polymerase and segments of HCMV polymerase that are important for foscarnet sensitivity has been solved.398 In the structure, the drug binds to highly conserved basic residues in a closed form of the enzyme that has not translocated to the next base on the template. The drug occupies the position of the beta and gamma phosphates of an incoming dNTP. The structure suggests that foscarnet stabilizes this untranslocated state, thus stalling the polymerase.
Because foscarnet is not a nucleoside analog, HSV tk mutants and HCMV UL97 mutants are not resistant to it. However, although foscarnet inhibits DNA polymerase by a mechanism that differs substantially from the nucleoside analogs, many pol mutants that are resistant to nucleoside analogs are resistant to foscarnet. Moreover, most foscarnet-resistant mutants are resistant to one or more nucleoside analogs (reviewed in 118). Therefore, there are patients with serious herpesvirus infections for whom there are no viable treatment options. This should be an impetus to further drug development.
Anti-HIV and HBV Polymerase Inhibitors
The retrovirus HIV and the hepadnavirus hepatitis B virus (HBV) replicate their genomes through DNA and RNA intermediates, respectively, using a DNA polymerase that can copy RNA. The HIV RT and the HBV DNA polymerase are thus essential for viral replication, and differ substantially from cellular polymerases, making them excellent targets for antivirals. The success of anti-herpesvirus nucleoside analogs provided a rationale for the development of nucleoside analogs active against HIV. Within 2 years of the identification of HIV as the etiologic agent of AIDS, a nucleoside analog, zidovudine (AZT), was shown to have antiviral activity in patients.395 Shortly thereafter AZT therapy was shown to have an impact on mortality in the short term.100 The success of AZT, but also its limitations, led to the development of other anti-HIV nucleoside analogs. These, in turn, provided an impetus for the development of nucleoside analogs against HBV, and, with the RT validated as a target, the nonnucleoside reverse transcriptase inhibitors (NNRTIs) against HIV.
Numerous nucleoside analogs have been approved to treat HIV (see Fig. 13.6). Many of the principles that govern their mechanisms of action and resistance are similar to those of the anti-herpesvirus nucleoside analogs. For the sake of brevity, this chapter focuses on only two anti-HIV nucleoside analogs,
zidovudine and lamivudine (which also is an anti-HBV drug), and compares and contrasts their mechanisms with each other and with the anti-herpesvirus drugs.
zidovudine and lamivudine (which also is an anti-HBV drug), and compares and contrasts their mechanisms with each other and with the anti-herpesvirus drugs.
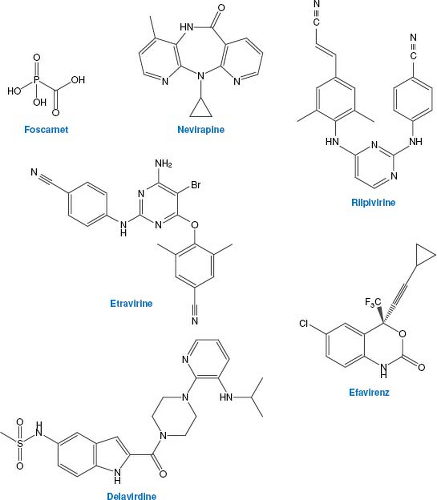 Figure 13.8. Nonnucleoside DNA polymerase inhibitors (foscarnet and nonnucleoside reverse transcriptase inhibitors [NNRTIs]). Foscarnet is a pyrophosphate analog that inhibits viral polymerases. It is approved for treatment of herpes simplex virus (HSV) and human cytomegalovirus (HCMV) infections that are resistant to other drugs. Nevirapine, delavirdine, efavirenz, etravirine, and rilpivirine, which do not mimic any known ligands of DNA polymerases, inhibit HIV-1 reverse transcriptase and are approved for treatment of HIV-1 in combination with other drugs. (Modified from Yeh RW, Coen DM. Pharmacology of viral infections. In: Golan DE, Tashjian AH, Jr., Armstrong EJ, et al., eds. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Third edition. Philadelphia: Lippincott Williams & Wilkins, 2012:649–673.) |
Zidovudine (AZT)
Zidovudine (azidothymidine, AZT) was synthesized years before the AIDS epidemic as a potential anticancer drug. It was first reported to have anti-HIV activity in 1985.255 Like the anti-herpesvirus drugs described earlier, AZT is a nucleoside analog with an altered sugar moiety. In this case, it is a thymine base attached to a sugar in which the normal 3′ hydroxyl has been replaced by an azido group (see Fig. 13.6). Indeed, all of the currently approved anti-HIV nucleoside analogs lack a 3′-hydroxyl or its equivalent. (This contrasts with penciclovir and ganciclovir.)
Unlike herpesviruses, HIV does not encode kinases that can phosphorylate nucleoside analogs. AZT is an excellent substrate for cellular thymidine kinase, which phosphorylates AZT to AZT-monophosphate.107 AZT-monophosphate is then converted to the diphosphate form by cellular thymidylate kinase and to the triphosphate form by cellular nucleoside diphosphate kinase.107 Therefore, unlike ACV and ganciclovir,
there is no selectivity at the activation step, and phosphorylated AZT accumulates in almost all dividing cells in the body, not just infected cells. Moreover, the activity of AZT and other anti-HIV nucleoside analogs can vary depending on the activity of cellular kinases in the infected cell. For example activated lymphocytes upregulate thymidine kinase, which is needed for DNA replication and cellular proliferation; thymidine analogs are thus more potent in activated than in quiescent CD4 lymphocytes.335
there is no selectivity at the activation step, and phosphorylated AZT accumulates in almost all dividing cells in the body, not just infected cells. Moreover, the activity of AZT and other anti-HIV nucleoside analogs can vary depending on the activity of cellular kinases in the infected cell. For example activated lymphocytes upregulate thymidine kinase, which is needed for DNA replication and cellular proliferation; thymidine analogs are thus more potent in activated than in quiescent CD4 lymphocytes.335
AZT-triphosphate (AZT-TP) is a substantially more potent inhibitor and better substrate of HIV RT than of the human DNA polymerases that have been tested.46,107,340 This biochemical selectivity is reflected in the degree of resistance of AZT-resistant mutants, which can be greater than 100-fold in some cell types.200 Like ACV triphosphate, AZT-TP lacks a 3′-hydroxyl, and is an obligatory chain terminator (see Fig. 13.6). The details of the inhibition of RT by AZT-TP are not entirely resolved, but given that AZT resistance acts on incorporated drug (see below), it is clear that the efficient incorporation of AZT-TP is crucial for its selectivity. Therefore, the potency of AZT and other nucleoside analogs depends on the levels of dNTPs that compete with the drug triphosphates for the RT.
Especially given that phosphorylated AZT accumulates in almost all dividing cells in the body, its toxicity is a serious clinical issue. In particular, AZT causes bone marrow suppression that manifests most commonly as neutropenia and anemia.308 AZT toxicity appears to be due not only to the effects of AZT-TP on cellular polymerases, but also to the effects of AZT-monophosphate, which is both a substrate for cellular thymidylate kinase, and an inhibitor of this essential enzyme.107 For a number of other anti-HIV nucleoside analogs, a key determinant of toxicity appears to be the mitochondrial DNA polymerase (DNA polymerase γ).43,209,235
The first AZT-resistant mutants of HIV were obtained from patients who had been treated with the drug for months.200 AZT-resistance mutations accumulate in the HIV pol gene that encodes RT24,53,187,203 (Fig. 13.9). Interestingly, four or more mutations are required to confer high-level resistance, but some of the mutations confer little or no resistance on their own.187,202,203 These latter mutations appear to be selected because they boost resistance by the other mutations or compensate for fitness costs of certain mutations. The requirement for multiple mutations for high-level resistance likely explains the relatively slow emergence of AZT resistance in patients and during selection in cell culture.111,201 Based on sequencing of virus isolated from patients following cessation of AZT therapy or in individuals newly infected with resistant virus, and of virus from replication competition experiments in vitro,
AZT-resistant mutants appear to be modestly less fit than wild-type virus.77,120,121,138,217
AZT-resistant mutants appear to be modestly less fit than wild-type virus.77,120,121,138,217
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


